{"title":"Targeting PRMT3 impairs methylation and oligomerization of HSP60 to boost anti-tumor immunity by activating cGAS/STING signaling","authors":"Yunxing Shi, Zongfeng Wu, Shaoru Liu, Dinglan Zuo, Yi Niu, Yuxiong Qiu, Liang Qiao, Wei He, Jiliang Qiu, Yunfei Yuan, Guocan Wang, Binkui Li","doi":"10.1038/s41467-024-52170-3","DOIUrl":null,"url":null,"abstract":"<p>Immune checkpoint blockade (ICB) has emerged as a promising therapeutic option for hepatocellular carcinoma (HCC), but resistance to ICB occurs and patient responses vary. Here, we uncover protein arginine methyltransferase 3 (PRMT3) as a driver for immunotherapy resistance in HCC. We show that PRMT3 expression is induced by ICB-activated T cells via an interferon-gamma (IFNγ)-STAT1 signaling pathway, and higher PRMT3 expression levels correlate with reduced numbers of tumor-infiltrating CD8<sup>+</sup> T cells and poorer response to ICB. Genetic depletion or pharmacological inhibition of PRMT3 elicits an influx of T cells into tumors and reduces tumor size in HCC mouse models. Mechanistically, PRMT3 methylates HSP60 at R446 to induce HSP60 oligomerization and maintain mitochondrial homeostasis. Targeting PRMT3-dependent HSP60 methylation disrupts mitochondrial integrity and increases mitochondrial DNA (mtDNA) leakage, which results in cGAS/STING-mediated anti-tumor immunity. Lastly, blocking PRMT3 functions synergize with PD-1 blockade in HCC mouse models. Our study thus identifies PRMT3 as a potential biomarker and therapeutic target to overcome immunotherapy resistance in HCC.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"52 1","pages":""},"PeriodicalIF":14.7000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-024-52170-3","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
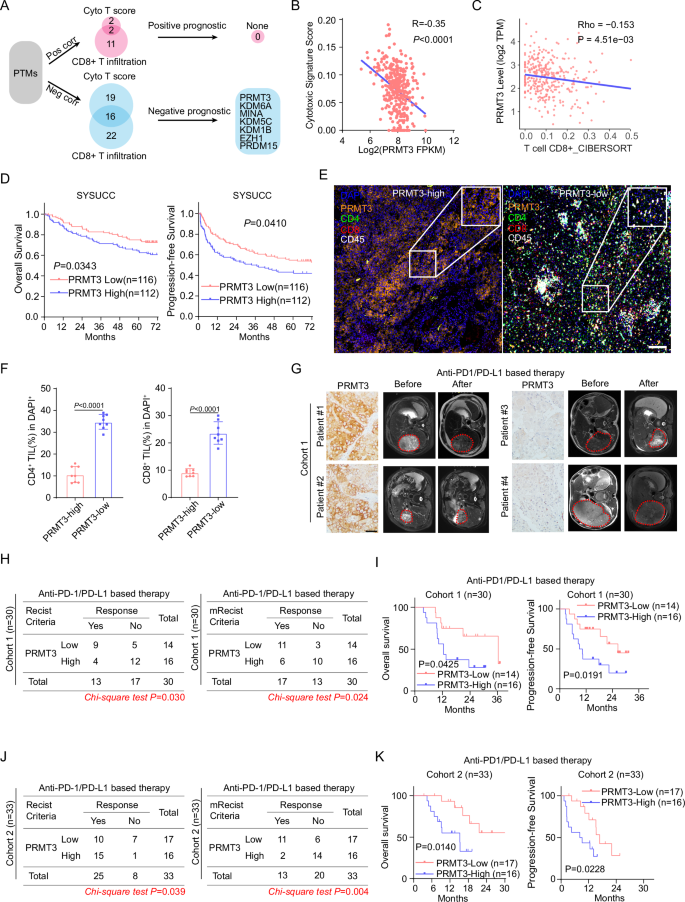
Immune checkpoint blockade (ICB) has emerged as a promising therapeutic option for hepatocellular carcinoma (HCC), but resistance to ICB occurs and patient responses vary. Here, we uncover protein arginine methyltransferase 3 (PRMT3) as a driver for immunotherapy resistance in HCC. We show that PRMT3 expression is induced by ICB-activated T cells via an interferon-gamma (IFNγ)-STAT1 signaling pathway, and higher PRMT3 expression levels correlate with reduced numbers of tumor-infiltrating CD8+ T cells and poorer response to ICB. Genetic depletion or pharmacological inhibition of PRMT3 elicits an influx of T cells into tumors and reduces tumor size in HCC mouse models. Mechanistically, PRMT3 methylates HSP60 at R446 to induce HSP60 oligomerization and maintain mitochondrial homeostasis. Targeting PRMT3-dependent HSP60 methylation disrupts mitochondrial integrity and increases mitochondrial DNA (mtDNA) leakage, which results in cGAS/STING-mediated anti-tumor immunity. Lastly, blocking PRMT3 functions synergize with PD-1 blockade in HCC mouse models. Our study thus identifies PRMT3 as a potential biomarker and therapeutic target to overcome immunotherapy resistance in HCC.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


