Nicholas Hopper, François Sidoroff, Juliette Cayer-Barrioz, Denis Mazuyer, Bo Chen and Wilfred T. Tysoe
{"title":"Modeling mechanochemistry: pressure dependence of Diels–Alder cycloaddition reaction kinetics†","authors":"Nicholas Hopper, François Sidoroff, Juliette Cayer-Barrioz, Denis Mazuyer, Bo Chen and Wilfred T. Tysoe","doi":"10.1039/D4MR00063C","DOIUrl":null,"url":null,"abstract":"<p >We analyze the effect of pressure on the Diels–Alder (D–A) dimerization reactions using Evans–Polanyi (E–P) theory, a thermodynamic analysis of the way in which a perturbation, in this case a hydrostatic pressure, modifies a reaction rate. Because it is a thermodynamic analysis, the results depend only on the volumes of the initial- and transition-state structures and not on the pathways between them. The volumes are calculated by enclosing the initial- and transition-state structures in a van der Waals' cocoon. Pressure is exerted by multiplying the van der Waals' radii by some factor without allowing the initial- and transition-state structures to relax. The influence of the surrounding solvent is included by using the extreme-pressure, polarizable-continuum method (XP-PCM). The approach is illustrated in detail using cyclopentadiene dimerization for which the rates have been independently measured by two groups. The analysis provides results that are in good agreement with those found experimentally for measurements made up to ∼0.3 GPa. The activation volumes of other D–A reactions are calculated in the same way and lead to good agreement for non-polar reactants, but less good agreement for polar ones. The pressure can also distort the initial- and transition-state structures, which can be calculated from the initial- and transition-state Hessians. A pressure-dependent distortion requires knowing the area over which the hydrostatic pressure acts. This is obtained using the Stearn–Eyring postulate that the activation volume is the product of an activation length and the area over which the stress acts. The activation length is obtained from quantum calculations of the difference in the distances between the diene and dienophile in the initial- and transition states. This provides only minor corrections to the results for routinely accessible hydrostatic pressures.</p>","PeriodicalId":101140,"journal":{"name":"RSC Mechanochemistry","volume":" 4","pages":" 402-412"},"PeriodicalIF":0.0000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/mr/d4mr00063c?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Mechanochemistry","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/mr/d4mr00063c","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
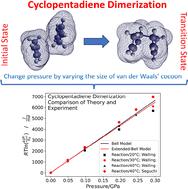
We analyze the effect of pressure on the Diels–Alder (D–A) dimerization reactions using Evans–Polanyi (E–P) theory, a thermodynamic analysis of the way in which a perturbation, in this case a hydrostatic pressure, modifies a reaction rate. Because it is a thermodynamic analysis, the results depend only on the volumes of the initial- and transition-state structures and not on the pathways between them. The volumes are calculated by enclosing the initial- and transition-state structures in a van der Waals' cocoon. Pressure is exerted by multiplying the van der Waals' radii by some factor without allowing the initial- and transition-state structures to relax. The influence of the surrounding solvent is included by using the extreme-pressure, polarizable-continuum method (XP-PCM). The approach is illustrated in detail using cyclopentadiene dimerization for which the rates have been independently measured by two groups. The analysis provides results that are in good agreement with those found experimentally for measurements made up to ∼0.3 GPa. The activation volumes of other D–A reactions are calculated in the same way and lead to good agreement for non-polar reactants, but less good agreement for polar ones. The pressure can also distort the initial- and transition-state structures, which can be calculated from the initial- and transition-state Hessians. A pressure-dependent distortion requires knowing the area over which the hydrostatic pressure acts. This is obtained using the Stearn–Eyring postulate that the activation volume is the product of an activation length and the area over which the stress acts. The activation length is obtained from quantum calculations of the difference in the distances between the diene and dienophile in the initial- and transition states. This provides only minor corrections to the results for routinely accessible hydrostatic pressures.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


