{"title":"MYH9-related inherited thrombocytopenia: the genetic spectrum, underlying mechanisms, clinical phenotypes, diagnosis, and management approaches","authors":"Kefeng Shen , Ting Chen , Min Xiao","doi":"10.1016/j.rpth.2024.102552","DOIUrl":null,"url":null,"abstract":"<div><p>Inherited thrombocytopenias have been considered exceedingly rare for a long time, but recent advances have facilitated diagnosis and greatly enabled the discovery of new causative genes. <em>MYH9</em>-related disease (<em>MYH9-</em>RD) represents one of the most frequent forms of inherited thrombocytopenia, usually presenting with nonspecific clinical manifestations, which renders it difficult to establish an accurate diagnosis. <em>MYH9-</em>RD is an autosomal dominant-inherited thrombocytopenia caused by deleterious variants in the <em>MYH9</em> gene encoding the heavy chain of nonmuscle myosin IIA. Patients with <em>MYH9</em>-RD usually present with thrombocytopenia and platelet macrocytosis at birth or in infancy, and most of them may develop one or more extrahematologic manifestations of progressive nephritis, sensorial hearing loss, presenile cataracts, and elevated liver enzymatic levels during childhood and adult life. Here, we have reviewed recent advances in the study of <em>MYH9</em>-RD, which aims to provide an updated and comprehensive summary of the current knowledge and improve our understanding of the genetic spectrum, underlying mechanisms, clinical phenotypes, diagnosis, and management approaches of this rare disease. Importantly, our goal is to enable physicians to better understand this rare disease and highlight the critical role of genetic etiologic analysis in ensuring accurate diagnosis, clinical management, and genetic counseling while avoiding ineffective and potentially harmful therapies for <em>MYH9</em>-RD patients.</p></div>","PeriodicalId":20893,"journal":{"name":"Research and Practice in Thrombosis and Haemostasis","volume":"8 6","pages":"Article 102552"},"PeriodicalIF":3.4000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2475037924002474/pdfft?md5=25f8cdaec23ba07ea71ddb4dc9a6f91c&pid=1-s2.0-S2475037924002474-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Research and Practice in Thrombosis and Haemostasis","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2475037924002474","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
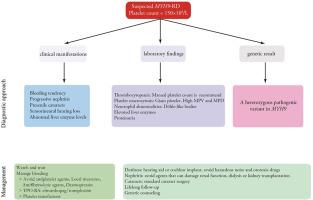
Inherited thrombocytopenias have been considered exceedingly rare for a long time, but recent advances have facilitated diagnosis and greatly enabled the discovery of new causative genes. MYH9-related disease (MYH9-RD) represents one of the most frequent forms of inherited thrombocytopenia, usually presenting with nonspecific clinical manifestations, which renders it difficult to establish an accurate diagnosis. MYH9-RD is an autosomal dominant-inherited thrombocytopenia caused by deleterious variants in the MYH9 gene encoding the heavy chain of nonmuscle myosin IIA. Patients with MYH9-RD usually present with thrombocytopenia and platelet macrocytosis at birth or in infancy, and most of them may develop one or more extrahematologic manifestations of progressive nephritis, sensorial hearing loss, presenile cataracts, and elevated liver enzymatic levels during childhood and adult life. Here, we have reviewed recent advances in the study of MYH9-RD, which aims to provide an updated and comprehensive summary of the current knowledge and improve our understanding of the genetic spectrum, underlying mechanisms, clinical phenotypes, diagnosis, and management approaches of this rare disease. Importantly, our goal is to enable physicians to better understand this rare disease and highlight the critical role of genetic etiologic analysis in ensuring accurate diagnosis, clinical management, and genetic counseling while avoiding ineffective and potentially harmful therapies for MYH9-RD patients.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


