Protein NMR assignment by isotope pattern recognition
IF 11.7
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
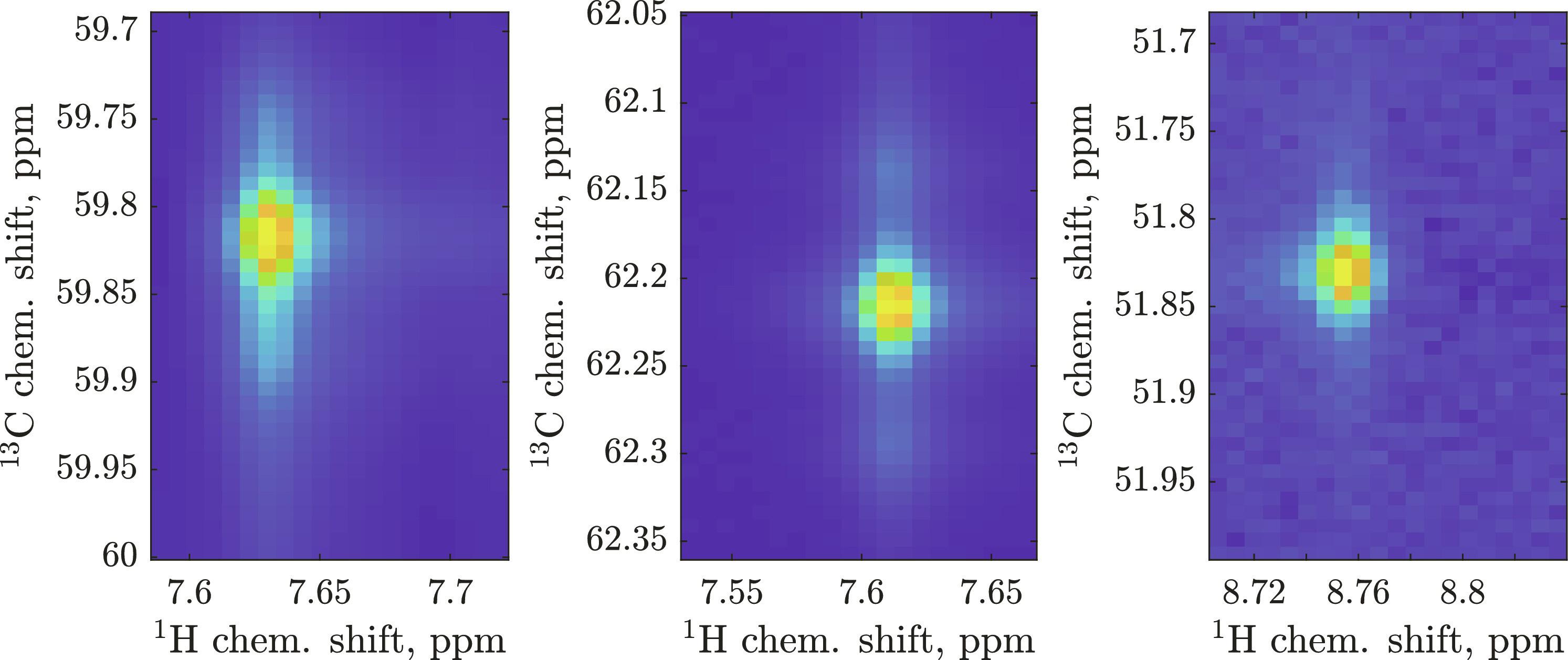
The current standard method for amino acid signal identification in protein NMR spectra is sequential assignment using triple-resonance experiments. Good software and elaborate heuristics exist, but the process remains laboriously manual. Machine learning does help, but its training databases need millions of samples that cover all relevant physics and every kind of instrumental artifact. In this communication, we offer a solution to this problem. We propose polyadic decompositions to store millions of simulated three-dimensional NMR spectra, on-the-fly generation of artifacts during training, a probabilistic way to incorporate prior and posterior information, and integration with the industry standard CcpNmr software framework. The resulting neural nets take [1H,13C] slices of mixed pyruvate–labeled HNCA spectra (different CA signal shapes for different residue types) and return an amino acid probability table. In combination with primary sequence information, backbones of common proteins (GB1, MBP, and INMT) are rapidly assigned from just the HNCA spectrum.
通过同位素模式识别进行蛋白质 NMR 分配。
蛋白质核磁共振光谱中氨基酸信号识别的现行标准方法是利用三重共振实验进行序列分配。虽然有很好的软件和精心设计的启发式方法,但这一过程仍然是费力的手工操作。机器学习确实有帮助,但它的训练数据库需要数百万个样本,涵盖所有相关物理和各种仪器工件。在这篇通讯中,我们提出了解决这一问题的方法。我们提出了存储数百万个模拟三维核磁共振波谱的多向分解、在训练过程中即时生成假象、纳入先验和后验信息的概率方法,以及与行业标准 CcpNmr 软件框架的集成。由此产生的神经网络能获取丙酮酸标记的混合 HNCA 图谱的[1H,13C]切片(不同残基类型的 CA 信号形状不同),并返回氨基酸概率表。结合主序列信息,仅通过 HNCA 图谱就能快速分配常见蛋白质(GB1、MBP 和 INMT)的骨架。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Science Advances
综合性期刊-综合性期刊
CiteScore
21.40
自引率
1.50%
发文量
1937
审稿时长
29 weeks
期刊介绍:
Science Advances, an open-access journal by AAAS, publishes impactful research in diverse scientific areas. It aims for fair, fast, and expert peer review, providing freely accessible research to readers. Led by distinguished scientists, the journal supports AAAS's mission by extending Science magazine's capacity to identify and promote significant advances. Evolving digital publishing technologies play a crucial role in advancing AAAS's global mission for science communication and benefitting humankind.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


