{"title":"Hereditary spastic paraplegia and extensive leukoencephalopathy: a case report of a unique phenotype associated with a GJB1/Cx32 p.Pro174Ser variant.","authors":"Haruko Nakamura, Hiroshi Doi, Yosuke Miyaji, Taishi Wada, Erisa Takahashi, Mikiko Tada, Hiromi Fukuda, Atsushi Fujita, Yuichi Higashiyama, Yuri Nagao, Kazue Kimura, Masaharu Hayashi, Kyoko Hoshino, Naomichi Matsumoto, Fumiaki Tanaka","doi":"10.1186/s12883-024-03823-9","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pathogenic variants in Gap junction protein beta 1 (GJB1), which encodes Connexin 32, are known to cause X-linked Charcot-Marie-Tooth disease (CMTX), the second most common form of CMT. CMTX presents with the following five central nervous systems (CNS) phenotypes: subclinical electrophysiological abnormalities, mild fixed abnormalities on neurological examination and/or imaging, transient CNS dysfunction, cognitive impairment, and persistent CNS manifestations.</p><p><strong>Case presentation: </strong>A 40-year-old Japanese male showed CNS symptoms, including nystagmus, prominent spastic paraplegia, and mild cerebellar ataxia, accompanied by subclinical peripheral neuropathy. Brain magnetic resonance imaging revealed hyperintensities in diffusion-weighted images of the white matter, particularly along the pyramidal tract, which had persisted since childhood. Nerve conduction assessment showed a mild decrease in motor conduction velocity, and auditory brainstem responses beyond wave II were absent. Peripheral and central conduction times in somatosensory evoked potentials elicited by stimulation of the median nerve were prolonged. Genetic analysis identified a hemizygous GJB1 variant, NM_000166.6:c.520C > T p.Pro174Ser.</p><p><strong>Conclusions: </strong>The patient in the case described here, with a GJB1 p.Pro174Ser variant, presented with a unique CNS-dominant phenotype, characterized by spastic paraplegia and persistent extensive leukoencephalopathy, rather than CMTX. Similar phenotypes have also been observed in patients with GJC2 and CLCN2 variants, likely because of the common function of these genes in regulating ion and water balance, which is essential for maintaining white matter function. CMTX should be considered within the spectrum of GJB1-related disorders, which can include patients with predominant CNS symptoms, some of which can potentially be classified as a new type of spastic paraplegia.</p>","PeriodicalId":9170,"journal":{"name":"BMC Neurology","volume":"24 1","pages":"310"},"PeriodicalIF":2.2000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11373513/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Neurology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12883-024-03823-9","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Pathogenic variants in Gap junction protein beta 1 (GJB1), which encodes Connexin 32, are known to cause X-linked Charcot-Marie-Tooth disease (CMTX), the second most common form of CMT. CMTX presents with the following five central nervous systems (CNS) phenotypes: subclinical electrophysiological abnormalities, mild fixed abnormalities on neurological examination and/or imaging, transient CNS dysfunction, cognitive impairment, and persistent CNS manifestations.
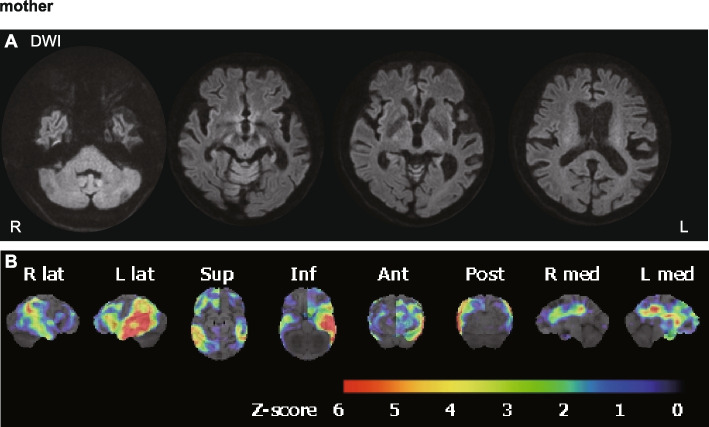
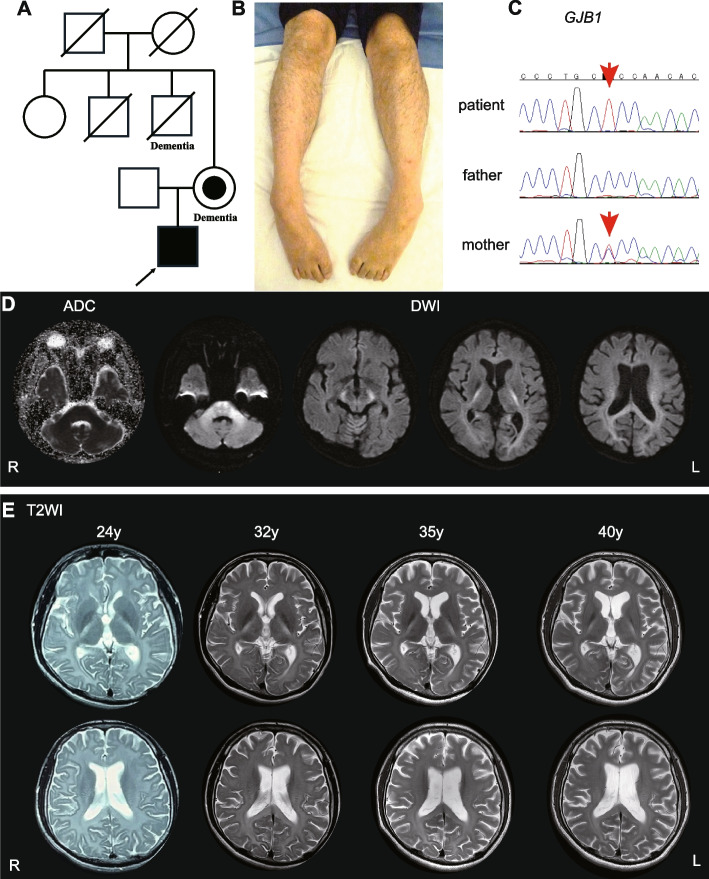
Case presentation: A 40-year-old Japanese male showed CNS symptoms, including nystagmus, prominent spastic paraplegia, and mild cerebellar ataxia, accompanied by subclinical peripheral neuropathy. Brain magnetic resonance imaging revealed hyperintensities in diffusion-weighted images of the white matter, particularly along the pyramidal tract, which had persisted since childhood. Nerve conduction assessment showed a mild decrease in motor conduction velocity, and auditory brainstem responses beyond wave II were absent. Peripheral and central conduction times in somatosensory evoked potentials elicited by stimulation of the median nerve were prolonged. Genetic analysis identified a hemizygous GJB1 variant, NM_000166.6:c.520C > T p.Pro174Ser.
Conclusions: The patient in the case described here, with a GJB1 p.Pro174Ser variant, presented with a unique CNS-dominant phenotype, characterized by spastic paraplegia and persistent extensive leukoencephalopathy, rather than CMTX. Similar phenotypes have also been observed in patients with GJC2 and CLCN2 variants, likely because of the common function of these genes in regulating ion and water balance, which is essential for maintaining white matter function. CMTX should be considered within the spectrum of GJB1-related disorders, which can include patients with predominant CNS symptoms, some of which can potentially be classified as a new type of spastic paraplegia.
期刊介绍:
BMC Neurology is an open access, peer-reviewed journal that considers articles on all aspects of the prevention, diagnosis and management of neurological disorders, as well as related molecular genetics, pathophysiology, and epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


