{"title":"S-nitrosoglutathione (GSNO) induces necroptotic cell death in K562 cells: Involvement of p73, TSC2 and SIRT1","authors":"Ayantika Sengupta, Subhamoy Chakraborty, Sanchita Biswas, Sourav Kumar Patra, Sanjay Ghosh","doi":"10.1016/j.cellsig.2024.111377","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Nitric oxide and Reactive Nitrogen Species are known to effect tumorigenicity. GSNO is one of the main NO carrying signalling moiety in cell. In the current study, we tried to delve into the effect of GSNO induced nitrosative stress in three different myelogenous leukemic K562, U937 and THP-1 cell lines.</p></div><div><h3>Method</h3><p>WST-8 assay was performed to investigate cell viability. RT-PCR and western-blot analysis were done to investigate mRNA and protein expression. Spectrophotometric and fluorimetric assays were done to investigate enzyme activities.</p></div><div><h3>Result</h3><p>We found that GSNO exposure led to reduced cell viability and the mode of cell death in K562 was non apoptotic in nature. GSNO promoted impaired autophagic flux and necroptosis. GSNO treatment heightened phosphorylation of AMPK and TSC2 and inhibited mTOR pathway. We observed increase in NAD<sup>+</sup>/ NADH ratio following GSNO treatment. Increase in both SIRT1 m-RNA and protein expression was observed. While total SIRT activity remained unaltered. GSNO increased tumor suppressor TAp73/ oncogenic ∆Np73 ratio in K562 cells which was correlated with cell mortality. Surprisingly, GSNO did not alter cellular redox status or redox associated protein expression. However, steep increase in total SNO and PSNO content was observed. Furthermore, inhibition of autophagy, AMPK phosphorylation or SIRT1 exacerbated the effect of GSNO. Altogether our work gives insights into GSNO mediated necroptotic event in K562 cells which can be excavated to develop NO based anticancer therapeutics.</p></div><div><h3>Conclusion</h3><p>Our data suggests that GSNO could induce necroptotic cell death in K562 through mitochondrial dysfunctionality and PTM of different cellular proteins.</p></div>","PeriodicalId":9902,"journal":{"name":"Cellular signalling","volume":"124 ","pages":"Article 111377"},"PeriodicalIF":4.4000,"publicationDate":"2024-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular signalling","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0898656824003450","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background
Nitric oxide and Reactive Nitrogen Species are known to effect tumorigenicity. GSNO is one of the main NO carrying signalling moiety in cell. In the current study, we tried to delve into the effect of GSNO induced nitrosative stress in three different myelogenous leukemic K562, U937 and THP-1 cell lines.
Method
WST-8 assay was performed to investigate cell viability. RT-PCR and western-blot analysis were done to investigate mRNA and protein expression. Spectrophotometric and fluorimetric assays were done to investigate enzyme activities.
Result
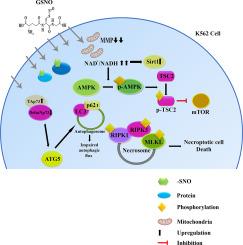
We found that GSNO exposure led to reduced cell viability and the mode of cell death in K562 was non apoptotic in nature. GSNO promoted impaired autophagic flux and necroptosis. GSNO treatment heightened phosphorylation of AMPK and TSC2 and inhibited mTOR pathway. We observed increase in NAD+/ NADH ratio following GSNO treatment. Increase in both SIRT1 m-RNA and protein expression was observed. While total SIRT activity remained unaltered. GSNO increased tumor suppressor TAp73/ oncogenic ∆Np73 ratio in K562 cells which was correlated with cell mortality. Surprisingly, GSNO did not alter cellular redox status or redox associated protein expression. However, steep increase in total SNO and PSNO content was observed. Furthermore, inhibition of autophagy, AMPK phosphorylation or SIRT1 exacerbated the effect of GSNO. Altogether our work gives insights into GSNO mediated necroptotic event in K562 cells which can be excavated to develop NO based anticancer therapeutics.
Conclusion
Our data suggests that GSNO could induce necroptotic cell death in K562 through mitochondrial dysfunctionality and PTM of different cellular proteins.
期刊介绍:
Cellular Signalling publishes original research describing fundamental and clinical findings on the mechanisms, actions and structural components of cellular signalling systems in vitro and in vivo.
Cellular Signalling aims at full length research papers defining signalling systems ranging from microorganisms to cells, tissues and higher organisms.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


