Emma M. Wade, Elizabeth A. Goodin, Tim Morgan, Stephana Pereira, Adele G. Woolley, Zandra A. Jenkins, Philip B. Daniel, Stephen P. Robertson
{"title":"The hinge-1 domain of Flna is not necessary for diverse physiological functions in mice","authors":"Emma M. Wade, Elizabeth A. Goodin, Tim Morgan, Stephana Pereira, Adele G. Woolley, Zandra A. Jenkins, Philip B. Daniel, Stephen P. Robertson","doi":"10.1111/eci.14308","DOIUrl":null,"url":null,"abstract":"<div>\n \n \n <section>\n \n <h3> Introduction</h3>\n \n <p>The filamins are cytoskeletal binding proteins that dynamically crosslink actin into orthogonal networks or bundle it into stress fibres. The domain structure of filamin proteins is very well characterised, with an N-terminal actin-binding region, followed by 24 immunoglobulin-like repeat units. The repeat domains are separated into distinct segments by two regions of low-complexity known as hinge-1 and hinge-2. The role of hinge-1 especially has been proposed to be essential for protein function as it provides flexibility to the otherwise rigid protein, and is a target for cleavage by calpain. Hinge-1 protects cells from otherwise destructive forces, and the products of calpain cleavage are involved in critical cellular signalling processes, such as survival during hypoxia. Pathogenic variants in <i>FLNA</i> encoding Filamin A, including those that remove the hinge-1 domain, cause a wide range of survivable developmental disorders. In contrast, complete loss of function of this gene is embryonic lethal in human and mouse.</p>\n </section>\n \n <section>\n \n <h3> Methods and Results</h3>\n \n <p>In this study, we show that removing filamin A hinge-1 from mouse (<i>Flna</i><sup><i>ΔH1</i></sup>), while preserving its expression level leads to no obvious developmental phenotype. Detailed characterisation of the skeletons of <i>Flna</i><sup><i>ΔH1</i></sup> mice showed no skeletal phenotype reminiscent of that found in the <i>FLNA-</i>causing skeletal dysplasia. Furthermore, nuclear functions of FLNA are maintained with loss of Filamin A hinge-1.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>We conclude that hinge-1 is dispensable for filamin A protein function during development over the murine lifespan.</p>\n </section>\n </div>","PeriodicalId":12013,"journal":{"name":"European Journal of Clinical Investigation","volume":"54 12","pages":""},"PeriodicalIF":4.4000,"publicationDate":"2024-08-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eci.14308","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"European Journal of Clinical Investigation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/eci.14308","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction
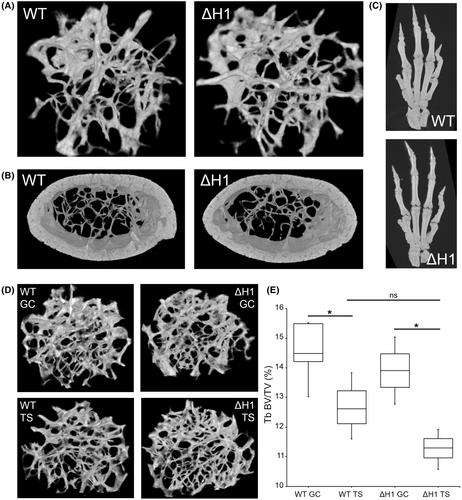
The filamins are cytoskeletal binding proteins that dynamically crosslink actin into orthogonal networks or bundle it into stress fibres. The domain structure of filamin proteins is very well characterised, with an N-terminal actin-binding region, followed by 24 immunoglobulin-like repeat units. The repeat domains are separated into distinct segments by two regions of low-complexity known as hinge-1 and hinge-2. The role of hinge-1 especially has been proposed to be essential for protein function as it provides flexibility to the otherwise rigid protein, and is a target for cleavage by calpain. Hinge-1 protects cells from otherwise destructive forces, and the products of calpain cleavage are involved in critical cellular signalling processes, such as survival during hypoxia. Pathogenic variants in FLNA encoding Filamin A, including those that remove the hinge-1 domain, cause a wide range of survivable developmental disorders. In contrast, complete loss of function of this gene is embryonic lethal in human and mouse.
Methods and Results
In this study, we show that removing filamin A hinge-1 from mouse (FlnaΔH1), while preserving its expression level leads to no obvious developmental phenotype. Detailed characterisation of the skeletons of FlnaΔH1 mice showed no skeletal phenotype reminiscent of that found in the FLNA-causing skeletal dysplasia. Furthermore, nuclear functions of FLNA are maintained with loss of Filamin A hinge-1.
Conclusion
We conclude that hinge-1 is dispensable for filamin A protein function during development over the murine lifespan.
期刊介绍:
EJCI considers any original contribution from the most sophisticated basic molecular sciences to applied clinical and translational research and evidence-based medicine across a broad range of subspecialties. The EJCI publishes reports of high-quality research that pertain to the genetic, molecular, cellular, or physiological basis of human biology and disease, as well as research that addresses prevalence, diagnosis, course, treatment, and prevention of disease. We are primarily interested in studies directly pertinent to humans, but submission of robust in vitro and animal work is also encouraged. Interdisciplinary work and research using innovative methods and combinations of laboratory, clinical, and epidemiological methodologies and techniques is of great interest to the journal. Several categories of manuscripts (for detailed description see below) are considered: editorials, original articles (also including randomized clinical trials, systematic reviews and meta-analyses), reviews (narrative reviews), opinion articles (including debates, perspectives and commentaries); and letters to the Editor.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


