Alexandra T Barth, Austin J Pyrch, Conor T McCormick, Evgeny O Danilov, Felix N Castellano
{"title":"Excited State Bond Homolysis of Vanadium(V) Photocatalysts for Alkoxy Radical Generation.","authors":"Alexandra T Barth, Austin J Pyrch, Conor T McCormick, Evgeny O Danilov, Felix N Castellano","doi":"10.1021/acs.jpca.4c04250","DOIUrl":null,"url":null,"abstract":"<p><p>Advancements in photocatalysis have transformed synthetic organic chemistry, using light as a powerful tool to drive selective chemical transformations. Recent approaches have focused on metal-halide ligand-to-metal charge transfer (LMCT) photoactivated bond homolysis reactions leveraged by earth-abundant elements to generate valuable synthons for radical-mediated cross-coupling reactions. Of recent utility, oxovanadium(V) LMCT photocatalysts exhibit selective alkoxy radical generation from aliphatic alcohols upon blue light (UVA) irradiation under mild conditions. The selective photochemical liberation of alkoxy radicals is valuable for applying late-stage fragmentation approaches in organic synthesis and depolymerization strategies for nonbiodegradable polymers. Steady-state and time-resolved spectroscopy were used to assign the electronic structure of three well-defined V(V) photocatalysts in their ground and excited states. We assign the excited state for this transformation at earth-abundant vanadium(V), interrogating the electronic structure using static UV-visible absorption, ultrafast transient absorption, and electron paramagnetic resonance spectroscopy coupled to computational approaches. These findings afford assignments of the short-lived excited state intermediates that dictate selective homolytic bond cleavage in metal alkoxides, illustrating the valuable insight gleaned from fundamental investigations of the molecular photochemistry responsible for light-escalated chemical transformations.</p>","PeriodicalId":2,"journal":{"name":"ACS Applied Bio Materials","volume":null,"pages":null},"PeriodicalIF":4.6000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Applied Bio Materials","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c04250","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/30 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"MATERIALS SCIENCE, BIOMATERIALS","Score":null,"Total":0}
引用次数: 0
Abstract
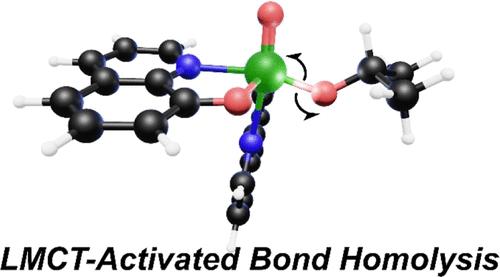
Advancements in photocatalysis have transformed synthetic organic chemistry, using light as a powerful tool to drive selective chemical transformations. Recent approaches have focused on metal-halide ligand-to-metal charge transfer (LMCT) photoactivated bond homolysis reactions leveraged by earth-abundant elements to generate valuable synthons for radical-mediated cross-coupling reactions. Of recent utility, oxovanadium(V) LMCT photocatalysts exhibit selective alkoxy radical generation from aliphatic alcohols upon blue light (UVA) irradiation under mild conditions. The selective photochemical liberation of alkoxy radicals is valuable for applying late-stage fragmentation approaches in organic synthesis and depolymerization strategies for nonbiodegradable polymers. Steady-state and time-resolved spectroscopy were used to assign the electronic structure of three well-defined V(V) photocatalysts in their ground and excited states. We assign the excited state for this transformation at earth-abundant vanadium(V), interrogating the electronic structure using static UV-visible absorption, ultrafast transient absorption, and electron paramagnetic resonance spectroscopy coupled to computational approaches. These findings afford assignments of the short-lived excited state intermediates that dictate selective homolytic bond cleavage in metal alkoxides, illustrating the valuable insight gleaned from fundamental investigations of the molecular photochemistry responsible for light-escalated chemical transformations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


