{"title":"Modeling Photodissociation: Quantum Dynamics Simulations of Methanol.","authors":"Léon L E Cigrang, Graham A Worth","doi":"10.1021/acs.jpca.4c03612","DOIUrl":null,"url":null,"abstract":"<p><p>A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG). Different initial excitation energies were simulated to investigate the dependence of the branching ratios on the electronic state being populated. A detailed mechanistic explanation is provided for the observed differences with respect to the excitation energy. Population of the lowest lying excited state of methanol leads to rapid hydroxyl hydrogen loss as the main dissociation channel. This is rationalized by the strongly dissociative nature of the PES cut along the O-H stretching coordinate, confirmed by the broad feature in the absorption spectrum. In contrast, more energetic excitations lead mainly to C-O bond breaking. Again, analysis of the diabatic surfaces offers a clear explanation in terms of the nature of the electronic states involved and the coupling between them. The type of calculations presented, as well as the subsequent analysis of the results, should be seen as a general workflow for the modeling of photochemical reactions.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11403662/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c03612","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
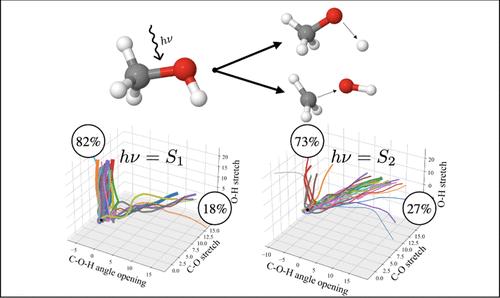
A comprehensive computational study of the gas-phase photodissociation dynamics of methanol is presented. Using a multiconfigurational active space based method (RASSCF) to obtain multidimensional potential energy surfaces (PESs) on-the-fly, direct quantum dynamics simulations were run using the variational multi-configurational Gaussian method (DD-vMCG). Different initial excitation energies were simulated to investigate the dependence of the branching ratios on the electronic state being populated. A detailed mechanistic explanation is provided for the observed differences with respect to the excitation energy. Population of the lowest lying excited state of methanol leads to rapid hydroxyl hydrogen loss as the main dissociation channel. This is rationalized by the strongly dissociative nature of the PES cut along the O-H stretching coordinate, confirmed by the broad feature in the absorption spectrum. In contrast, more energetic excitations lead mainly to C-O bond breaking. Again, analysis of the diabatic surfaces offers a clear explanation in terms of the nature of the electronic states involved and the coupling between them. The type of calculations presented, as well as the subsequent analysis of the results, should be seen as a general workflow for the modeling of photochemical reactions.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


