A methylation-related lncRNA-based prediction model in lung adenocarcinomas
Abstract
Background
The collaboration between methylation and the lung adenocarcinoma (LUAD) occurrence and development is closes. Long noncoding RNA (lncRNA), as a regulatory factor of various biological functions, can be used for cancer diagnosis. Our study aimed to construct a robust methylation-related lncRNA signature of LUAD.
Methods
In the Cancer Genome Atlas (TCGA) dataset, we download the RNA expression data and clinical information of LUAD cases. To develop the best prognostic signature based on methylation-related lncRNAs, Cox regression analyses were utilized. Using Kaplan–Meier analysis, overall survival rates were compared between risk category included both low- and high-risk patients. To categorize genes according to their functional significance, GSEA (Subramanian et al, 2005) was used. Single-sample gene set enrichment analysis (ssGSEA) was used to further reveal the potential molecular mechanism of the methylation-related lncRNA prognostic model in immune infiltration. Using TRLnc (http://www.licpathway.net/TRlnc) and lncRNASNP to analyse the SNP sites and TRLnc of these 18 lncRNAs. LncSEA website was used to analyse 18 lncRNA in the process of tumour development and development. Go was used to analyse the enriched pathways enriched by TFs (transcription factors), Cerna networks, and proteins bound to each other of these 18 lncRNAs. The ‘prophetic’ package was used to analyse the value of this prognostic model in guiding personalized immunotherapy.
Results
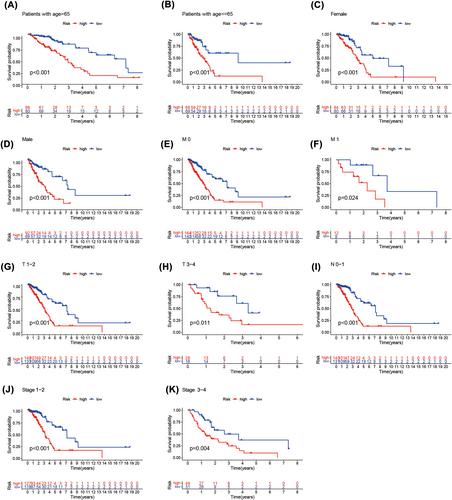
In this study, we identified 18 methylation-related lncRNAs (AP002761.1, AL118558.3, CH17-340M24.3, AL353150.1, AC004687.1, LINC00996, AF186192.1, HSPC324, AC087752.3, FAM30A, AC106047.1, AC026355.1, ABALON, LINC01843, AL606489.1, NKILA, AP001453.2, GSEC) to establish a methylation-related lncRNA signature that can detect patients prognosis in LUAD. The enriched pathways enriched by proteins interacting with 18 lncRNAs are mainly EMT, hypoxia, stemness and proliferation, among which LINC00996 and AF186192.1 are regulated by multiple tumour associated transcription factors, such as TP53 and TP63, and fam30a and mRNA form a Cerna network. There are 2319 SNP loci in LINC00996, 36 of which are risk SNP loci and 205 SNP loci in af186192.1; AF186192.1 affects 95 conserved miRNAs and 123 non-conserved miRNAs, promotes the binding of 149 pairs of miRNAs: lncRNAs and inhibits the binding of 95 pairs of miRNAs: lncRNAs. The ROC curve demonstrated that the established methylation-related lncRNA signature was more effective in predicting the prognosis of patients in LUAD than the clinicopathological parameters. Our research has confirmed that patients in the high-risk group which was separated by the risk score model based on methylation-related lncRNA had shorter OS. According to GSEA, the high-risk group had a predominantly tumour- and immune-related pathway enrichment. A significant association was shown by ssGSEA between predictive signature and immune status in LUAD patients. In addition, principal component analysis (PCA) demonstrated the prognostic and predictive value of our signature. The correlation between the predictive signature of methylation-related lncRNA and IC50 of conventional chemotherapy drugs can provide personalized chemotherapy regimens for LUAD patients. Methylation-related lncRNA signature can effectively predict DFS of patients in LUAD.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


