Leopoldo Mejía, Sandeep Sharma, Roi Baer, Garnet Kin-Lic Chan, Eran Rabani
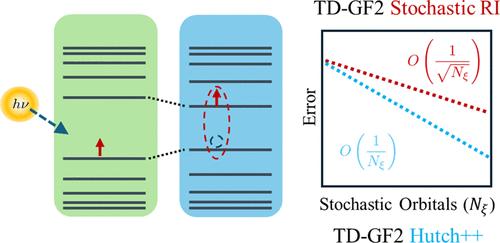
{"title":"Convergence Analysis of the Stochastic Resolution of Identity: Comparing Hutchinson to Hutch++ for the Second-Order Green's Function.","authors":"Leopoldo Mejía, Sandeep Sharma, Roi Baer, Garnet Kin-Lic Chan, Eran Rabani","doi":"10.1021/acs.jctc.4c00862","DOIUrl":null,"url":null,"abstract":"<p><p>Stochastic orbital techniques offer reduced computational scaling and memory requirements to describe ground and excited states at the cost of introducing controlled statistical errors. Such techniques often rely on two basic operations, stochastic trace estimation and stochastic resolution of identity, both of which lead to statistical errors that scale with the number of stochastic realizations (<i>N</i><sub>ξ</sub>) as <math><msqrt><mrow><msubsup><mi>N</mi><mi>ξ</mi><mrow><mo>-</mo><mn>1</mn></mrow></msubsup></mrow></msqrt></math>. Reducing the statistical errors without significantly increasing <i>N</i><sub>ξ</sub> has been challenging and is central to the development of efficient and accurate stochastic algorithms. In this work, we build upon recent progress made to improve stochastic trace estimation based on the ubiquitous Hutchinson's algorithm and propose a two-step approach for the stochastic resolution of identity, in the spirit of the Hutch++ method. Our approach is based on employing a randomized low-rank approximation followed by a residual calculation, resulting in statistical errors that scale much better than <math><msqrt><mrow><msubsup><mi>N</mi><mi>ξ</mi><mrow><mo>-</mo><mn>1</mn></mrow></msubsup></mrow></msqrt></math>. We implement the approach within the second-order Born approximation for the self-energy in the computation of neutral excitations and discuss three different low-rank approximations for the two-body Coulomb integrals. Tests on a series of hydrogen dimer chains with varying lengths demonstrate that the Hutch++-like approximations are computationally more efficient than both deterministic and purely stochastic (Hutchinson) approaches for low error thresholds and intermediate system sizes. Notably, for arbitrarily large systems, the Hutchinson-like approximation outperforms both deterministic and Hutch++-like methods.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00862","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
Stochastic orbital techniques offer reduced computational scaling and memory requirements to describe ground and excited states at the cost of introducing controlled statistical errors. Such techniques often rely on two basic operations, stochastic trace estimation and stochastic resolution of identity, both of which lead to statistical errors that scale with the number of stochastic realizations (Nξ) as . Reducing the statistical errors without significantly increasing Nξ has been challenging and is central to the development of efficient and accurate stochastic algorithms. In this work, we build upon recent progress made to improve stochastic trace estimation based on the ubiquitous Hutchinson's algorithm and propose a two-step approach for the stochastic resolution of identity, in the spirit of the Hutch++ method. Our approach is based on employing a randomized low-rank approximation followed by a residual calculation, resulting in statistical errors that scale much better than . We implement the approach within the second-order Born approximation for the self-energy in the computation of neutral excitations and discuss three different low-rank approximations for the two-body Coulomb integrals. Tests on a series of hydrogen dimer chains with varying lengths demonstrate that the Hutch++-like approximations are computationally more efficient than both deterministic and purely stochastic (Hutchinson) approaches for low error thresholds and intermediate system sizes. Notably, for arbitrarily large systems, the Hutchinson-like approximation outperforms both deterministic and Hutch++-like methods.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


