Justin L. Engel, Xiao Zhang, Mingming Wu, Yan Wang, Jose Espejo Valle-Inclán, Qing Hu, Kidist S. Woldehawariat, Mathijs A. Sanders, Agata Smogorzewska, Jin Chen, Isidro Cortés-Ciriano, Roger S. Lo, Peter Ly
{"title":"The Fanconi anemia pathway induces chromothripsis and ecDNA-driven cancer drug resistance","authors":"Justin L. Engel, Xiao Zhang, Mingming Wu, Yan Wang, Jose Espejo Valle-Inclán, Qing Hu, Kidist S. Woldehawariat, Mathijs A. Sanders, Agata Smogorzewska, Jin Chen, Isidro Cortés-Ciriano, Roger S. Lo, Peter Ly","doi":"10.1016/j.cell.2024.08.001","DOIUrl":null,"url":null,"abstract":"<p>Chromothripsis describes the catastrophic shattering of mis-segregated chromosomes trapped within micronuclei. Although micronuclei accumulate DNA double-strand breaks and replication defects throughout interphase, how chromosomes undergo shattering remains unresolved. Using CRISPR-Cas9 screens, we identify a non-canonical role of the Fanconi anemia (FA) pathway as a driver of chromothripsis. Inactivation of the FA pathway suppresses chromosome shattering during mitosis without impacting interphase-associated defects within micronuclei. Mono-ubiquitination of FANCI-FANCD2 by the FA core complex promotes its mitotic engagement with under-replicated micronuclear chromosomes. The structure-selective SLX4-XPF-ERCC1 endonuclease subsequently induces large-scale nucleolytic cleavage of persistent DNA replication intermediates, which stimulates POLD3-dependent mitotic DNA synthesis to prime shattered fragments for reassembly in the ensuing cell cycle. Notably, FA-pathway-induced chromothripsis generates complex genomic rearrangements and extrachromosomal DNA that confer acquired resistance to anti-cancer therapies. Our findings demonstrate how pathological activation of a central DNA repair mechanism paradoxically triggers cancer genome evolution through chromothripsis.</p>","PeriodicalId":9656,"journal":{"name":"Cell","volume":"6 1","pages":""},"PeriodicalIF":45.5000,"publicationDate":"2024-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.cell.2024.08.001","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
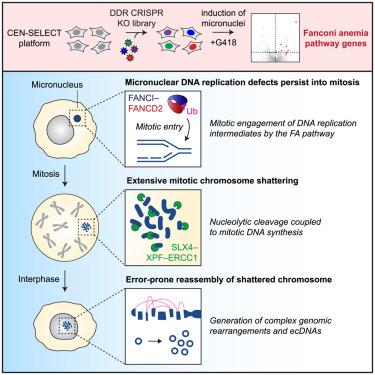
Chromothripsis describes the catastrophic shattering of mis-segregated chromosomes trapped within micronuclei. Although micronuclei accumulate DNA double-strand breaks and replication defects throughout interphase, how chromosomes undergo shattering remains unresolved. Using CRISPR-Cas9 screens, we identify a non-canonical role of the Fanconi anemia (FA) pathway as a driver of chromothripsis. Inactivation of the FA pathway suppresses chromosome shattering during mitosis without impacting interphase-associated defects within micronuclei. Mono-ubiquitination of FANCI-FANCD2 by the FA core complex promotes its mitotic engagement with under-replicated micronuclear chromosomes. The structure-selective SLX4-XPF-ERCC1 endonuclease subsequently induces large-scale nucleolytic cleavage of persistent DNA replication intermediates, which stimulates POLD3-dependent mitotic DNA synthesis to prime shattered fragments for reassembly in the ensuing cell cycle. Notably, FA-pathway-induced chromothripsis generates complex genomic rearrangements and extrachromosomal DNA that confer acquired resistance to anti-cancer therapies. Our findings demonstrate how pathological activation of a central DNA repair mechanism paradoxically triggers cancer genome evolution through chromothripsis.
染色体破碎(Chromothripsis)是指被困在微核中的错误分离的染色体发生灾难性破碎。虽然微核在整个间期都会积累 DNA 双链断裂和复制缺陷,但染色体是如何发生破碎的仍未解决。通过 CRISPR-Cas9 筛选,我们确定了范可尼贫血症(FA)通路作为染色体破碎驱动因素的非经典作用。FA通路的失活抑制了有丝分裂过程中的染色体破碎,但不会影响微核内的间期相关缺陷。FA核心复合物对FANCI-FANCD2的单泛素化促进了FANCI-FANCD2与复制不足的小核染色体的有丝分裂啮合。随后,结构选择性 SLX4-XPF-ERCC1 内切酶诱导对持续存在的 DNA 复制中间体进行大规模核溶解切割,从而刺激 POLD3 依赖性有丝分裂 DNA 合成,使破碎的片段在随后的细胞周期中重新组合。值得注意的是,FA 途径诱导的染色体分裂会产生复杂的基因组重排和染色体外 DNA,从而对抗癌疗法产生获得性抗性。我们的研究结果表明,病理激活中心DNA修复机制是如何通过染色体三分裂矛盾地引发癌症基因组进化的。
期刊介绍:
Cells is an international, peer-reviewed, open access journal that focuses on cell biology, molecular biology, and biophysics. It is affiliated with several societies, including the Spanish Society for Biochemistry and Molecular Biology (SEBBM), Nordic Autophagy Society (NAS), Spanish Society of Hematology and Hemotherapy (SEHH), and Society for Regenerative Medicine (Russian Federation) (RPO).
The journal publishes research findings of significant importance in various areas of experimental biology, such as cell biology, molecular biology, neuroscience, immunology, virology, microbiology, cancer, human genetics, systems biology, signaling, and disease mechanisms and therapeutics. The primary criterion for considering papers is whether the results contribute to significant conceptual advances or raise thought-provoking questions and hypotheses related to interesting and important biological inquiries.
In addition to primary research articles presented in four formats, Cells also features review and opinion articles in its "leading edge" section, discussing recent research advancements and topics of interest to its wide readership.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


