Abbas Agaimy, Andres M Acosta, Liang Cheng, Katrina Collins, Eddie Fridman, Christoph Schubart, Sean R Williamson, Arndt Hartmann, Kiril Trpkov
{"title":"TFE3-rearranged nonmelanotic renal PEComa: a case series expanding their phenotypic and fusion landscape","authors":"Abbas Agaimy, Andres M Acosta, Liang Cheng, Katrina Collins, Eddie Fridman, Christoph Schubart, Sean R Williamson, Arndt Hartmann, Kiril Trpkov","doi":"10.1111/his.15304","DOIUrl":null,"url":null,"abstract":"<div>\n \n <section>\n \n <h3> Aims</h3>\n \n <p>A subset of exceptionally rare primary renal perivascular epithelioid cell tumours (PEComas) that harbour Xp11.2 translocation have been reported, but no larger series devoted to this topic have been published.</p>\n </section>\n \n <section>\n \n <h3> Methods and Results</h3>\n \n <p>We describe the clinicopathological and molecular features of 10 renal PEComas, collected from our routine and consultation files. There were five female and five male patients aged 14–65 (median: 32 years). One patient had a history of childhood neuroblastoma, but no patients were known to have a tuberous sclerosis complex or other hereditary disorder. Complete surgical excision was the treatment for all patients. The available follow-up in five patients indicated a favourable outcome in 4/5 cases. Tumour size ranged from 2.8 to 15.2 cm (median, 5.2 cm). Immunohistochemistry revealed consistently strong TFE3 expression in all tumours, whereas PAX8 and keratin cocktails were uniformly negative. Other positive markers included HMB45 (7/9 tumours), CathepsinK (7/9 tumours), and CD117 (KIT) (3/5 tumours). <i>TFE3</i> rearrangements were detected in 8/9 tumours (by targeted RNA sequencing in seven and by FISH in one). The identified fusion partners included <i>SFPQ</i> (<i>n</i> = 2) and one tumour each with <i>ASPSCR1</i>, <i>ZC3H4</i>, <i>MED15</i>, <i>RBMX</i>, and <i>PRCC</i>. One tumour that lacked <i>TFE3</i> rearrangement by next-generation sequencing (NGS) and fluorescence <i>in situ</i> hybridization (FISH) revealed a large intrachromosomal deletion involving <i>PKD1</i> and <i>TSC2</i> by DNA-based NGS.</p>\n </section>\n \n <section>\n \n <h3> Conclusion</h3>\n \n <p>This study highlights the morphologic and genetic diversity of <i>TFE3</i>-rearranged primary renal PEComas and underlines the value of surrogate TFE3 immunohistochemistry in identifying them. The lack of PAX8 and keratin expression represents the mainstay for distinguishing these tumours from MiTF-associated renal cell carcinomas. In addition, we report rare (<i>ZC3H4</i>, <i>RBMX</i>) and novel (<i>MED15</i>) <i>TFE3</i> fusion partners in PEComa.</p>\n </section>\n </div>","PeriodicalId":13219,"journal":{"name":"Histopathology","volume":"85 5","pages":"783-793"},"PeriodicalIF":3.9000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/his.15304","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Histopathology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/his.15304","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Aims
A subset of exceptionally rare primary renal perivascular epithelioid cell tumours (PEComas) that harbour Xp11.2 translocation have been reported, but no larger series devoted to this topic have been published.
Methods and Results
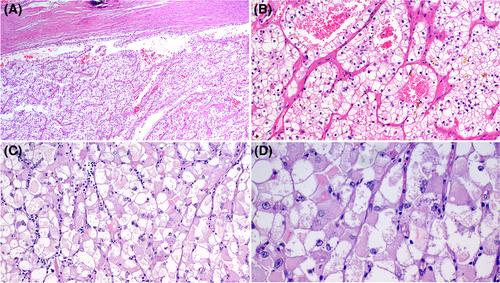
We describe the clinicopathological and molecular features of 10 renal PEComas, collected from our routine and consultation files. There were five female and five male patients aged 14–65 (median: 32 years). One patient had a history of childhood neuroblastoma, but no patients were known to have a tuberous sclerosis complex or other hereditary disorder. Complete surgical excision was the treatment for all patients. The available follow-up in five patients indicated a favourable outcome in 4/5 cases. Tumour size ranged from 2.8 to 15.2 cm (median, 5.2 cm). Immunohistochemistry revealed consistently strong TFE3 expression in all tumours, whereas PAX8 and keratin cocktails were uniformly negative. Other positive markers included HMB45 (7/9 tumours), CathepsinK (7/9 tumours), and CD117 (KIT) (3/5 tumours). TFE3 rearrangements were detected in 8/9 tumours (by targeted RNA sequencing in seven and by FISH in one). The identified fusion partners included SFPQ (n = 2) and one tumour each with ASPSCR1, ZC3H4, MED15, RBMX, and PRCC. One tumour that lacked TFE3 rearrangement by next-generation sequencing (NGS) and fluorescence in situ hybridization (FISH) revealed a large intrachromosomal deletion involving PKD1 and TSC2 by DNA-based NGS.
Conclusion
This study highlights the morphologic and genetic diversity of TFE3-rearranged primary renal PEComas and underlines the value of surrogate TFE3 immunohistochemistry in identifying them. The lack of PAX8 and keratin expression represents the mainstay for distinguishing these tumours from MiTF-associated renal cell carcinomas. In addition, we report rare (ZC3H4, RBMX) and novel (MED15) TFE3 fusion partners in PEComa.
期刊介绍:
Histopathology is an international journal intended to be of practical value to surgical and diagnostic histopathologists, and to investigators of human disease who employ histopathological methods. Our primary purpose is to publish advances in pathology, in particular those applicable to clinical practice and contributing to the better understanding of human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


