Neural-network-based molecular dynamics simulations reveal that proton transport in water is doubly gated by sequential hydrogen-bond exchange
IF 19.2
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
The transport of excess protons in water is central to acid–base chemistry, biochemistry and energy production. However, elucidating its mechanism has been challenging. Recent nonlinear vibrational spectroscopy experiments could not be explained by existing models. Here we use both vibrational spectroscopy calculations and neural-network-based molecular dynamics simulations that account for nuclear quantum effects for all atoms to determine the proton transport mechanism. Our simulations reveal an equilibrium between two stable proton-localized structures with distinct Eigen-like and Zundel-like hydrogen-bond motifs. Proton transport follows a three-step mechanism gated by two successive hydrogen-bond exchanges: the first reduces the proton-acceptor water coordination, leading to proton transfer, and the second, the rate-limiting step, prevents rapid back-transfer by increasing the proton-donor coordination. This sequential mechanism is consistent with experimental characterizations of proton diffusion, explaining the low activation energy and the prolonged intermediate lifetimes in vibrational spectroscopy. These results are crucial for understanding proton dynamics in biochemical and technological systems. Recent vibrational spectroscopy experiments suggested that excess proton transport in water is more complex than previously thought. Now the proton transport mechanism has been explored using neural-network-based simulations and related to experimental measurements through vibrational spectra calculations. It was observed to be a three-step process gated by two successive hydrogen-bond exchanges.
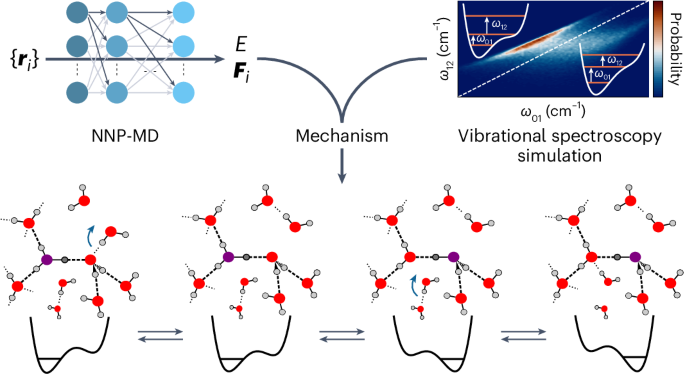
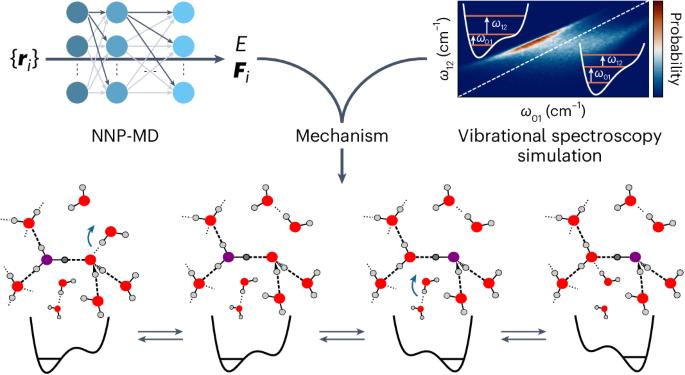
基于神经网络的分子动力学模拟揭示了质子在水中的迁移是由连续的氢键交换双重控制的
过量质子在水中的迁移是酸碱化学、生物化学和能源生产的核心。然而,阐明其机理一直是一项挑战。现有模型无法解释最近的非线性振动光谱实验。在这里,我们利用振动光谱计算和基于神经网络的分子动力学模拟(考虑到所有原子的核量子效应)来确定质子传输机制。我们的模拟揭示了两种稳定的质子定位结构之间的平衡,这两种结构具有不同的类 Eigen 和类 Zundel 氢键图案。质子迁移遵循一个由两次连续氢键交换驱动的三步机制:第一步降低质子受体水配位,导致质子转移;第二步,即限制速率的一步,通过增加质子供体配位防止快速反向转移。这种顺序机制与质子扩散的实验特征一致,解释了振动光谱中的低活化能和中间寿命延长的原因。这些结果对于理解生化和技术系统中的质子动力学至关重要。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature chemistry
化学-化学综合
CiteScore
29.60
自引率
1.40%
发文量
226
审稿时长
1.7 months
期刊介绍:
Nature Chemistry is a monthly journal that publishes groundbreaking and significant research in all areas of chemistry. It covers traditional subjects such as analytical, inorganic, organic, and physical chemistry, as well as a wide range of other topics including catalysis, computational and theoretical chemistry, and environmental chemistry.
The journal also features interdisciplinary research at the interface of chemistry with biology, materials science, nanotechnology, and physics. Manuscripts detailing such multidisciplinary work are encouraged, as long as the central theme pertains to chemistry.
Aside from primary research, Nature Chemistry publishes review articles, news and views, research highlights from other journals, commentaries, book reviews, correspondence, and analysis of the broader chemical landscape. It also addresses crucial issues related to education, funding, policy, intellectual property, and the societal impact of chemistry.
Nature Chemistry is dedicated to ensuring the highest standards of original research through a fair and rigorous review process. It offers authors maximum visibility for their papers, access to a broad readership, exceptional copy editing and production standards, rapid publication, and independence from academic societies and other vested interests.
Overall, Nature Chemistry aims to be the authoritative voice of the global chemical community.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


