Variants in IGLL1 cause a broad phenotype from agammaglobulinemia to transient hypogammaglobulinemia
IF 11.2
1区 医学
Q1 ALLERGY
引用次数: 0
Abstract
Background
Agammaglobulinemia due to variants in IGLL1 has traditionally been considered an exceedingly rare form of severe B-cell deficiency, with only 8 documented cases in the literature. Surprisingly, the first agammaglobulinemic patient identified by newborn screening (NBS) through quantification of kappa-deleting recombination excision circles harbored variants in IGLL1.
Objective
We comprehensively reviewed clinical and immunologic findings of patients with B-cell deficiency attributed to variants in IGLL1.
Methods
NBS programs reporting the use of kappa-deleting recombination excision circle assays, the European Society for Immunodeficiencies Registry, and authors of published reports featuring patients with B-cell deficiency linked to IGLL1 variants were contacted. Only patients with (likely) pathogenic variants, reduced CD19+ counts, and no alternative diagnosis were included.
Results
The study included 13 patients identified through NBS, 2 clinically diagnosed patients, and 2 asymptomatic siblings. All had severely reduced CD19+ B cells (< 0.1 × 109/L) at first evaluation, yet subsequent follow-up assessments indicated residual immunoglobulin production. Specific antibody responses to vaccine antigens varied, with a predominant reduction observed during infancy. Clinical outcomes were favorable with IgG substitution. Two patients successfully discontinued substitution therapy without developing susceptibility to infections and while maintaining immunoglobulin levels. The pooled incidence of homozygous or compound heterozygous pathogenic IGLL1 variants identified by NBS in Austria, Czechia, and Switzerland was 1.3:100,000, almost double of X-linked agammaglobulinemia.
Conclusion
B-cell deficiency resulting from IGLL1 variants appears to be more prevalent than initially believed. Despite markedly low B-cell counts, the clinical course in some patients may be milder than reported in the literature so far.
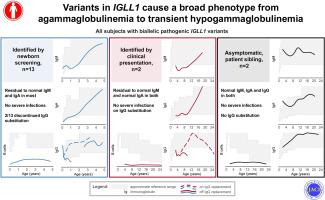
IGLL1 的变异可导致从阿加球蛋白血症到一过性低阿加球蛋白血症等多种表型。
背景:传统上,IGLL1变异导致的农抗球蛋白血症被认为是一种极为罕见的严重B细胞缺乏症,文献中仅有8例记录在案。令人惊讶的是,第一例通过新生儿筛查(NBS)定量检测卡帕缺失重组切除圈发现的丙种球蛋白血症患者携带 IGLL1 变异:全面概述因 IGLL1 变异而导致 B 细胞缺乏症患者的临床和免疫学结果:方法:我们联系了使用卡帕缺失重组切除圈检测法进行报告的 NBS 项目、欧洲免疫缺陷协会注册中心(European Society for Immunodeficiencies Registry),以及已发表的与 IGLL1 变异有关的 B 细胞缺乏症患者报告的作者。只有具有(可能)致病变体、CD19+计数减少且无其他诊断结果的患者才被纳入研究范围:研究包括 13 名通过 NBS 确定的患者、两名临床诊断患者和两名无症状的兄弟姐妹。首次评估时,所有患者的 CD19+ B 细胞均严重减少(< 0.1×109/L),但随后的随访结果显示仍有免疫球蛋白产生。对疫苗抗原的特异性抗体反应各不相同,主要是在婴儿期出现减少。免疫球蛋白 G 替代的临床效果良好。两名患者成功终止了替代治疗,没有出现感染易感性并保持了免疫球蛋白水平。在奥地利、捷克和瑞士,通过 NBS 发现的同卵或复合杂合致病性 IGLL1 变体的总发病率为 1.3:100´000,几乎是 X 连锁丙种球蛋白血症的两倍:结论:IGLL1 变体导致的 B 细胞缺乏症似乎比最初认为的更为普遍。结论:IGLL1 变体导致的 B 细胞缺乏症似乎比最初认为的更为普遍。尽管 B 细胞计数明显偏低,但一些患者的临床病程可能比迄今为止文献报道的要轻。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
25.90
自引率
7.70%
发文量
1302
审稿时长
38 days
期刊介绍:
The Journal of Allergy and Clinical Immunology is a prestigious publication that features groundbreaking research in the fields of Allergy, Asthma, and Immunology. This influential journal publishes high-impact research papers that explore various topics, including asthma, food allergy, allergic rhinitis, atopic dermatitis, primary immune deficiencies, occupational and environmental allergy, and other allergic and immunologic diseases. The articles not only report on clinical trials and mechanistic studies but also provide insights into novel therapies, underlying mechanisms, and important discoveries that contribute to our understanding of these diseases. By sharing this valuable information, the journal aims to enhance the diagnosis and management of patients in the future.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


