Xingying Zhu, Wai W Cheung, Aihua Zhang, Guixia Ding
{"title":"Mutation Characteristics of Primary Hyperoxaluria in the Chinese Population and Current International Diagnosis and Treatment Status.","authors":"Xingying Zhu, Wai W Cheung, Aihua Zhang, Guixia Ding","doi":"10.1159/000539516","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary hyperoxaluria (PH) is a rare autosomal recessive disorder, mainly due to the increase in endogenous oxalate production, causing a series of clinical features such as kidney stones, nephrocalcinosis, progressive impairment of renal function, and systemic oxalosis. There are three common genetic causes of glycolate metabolism anomalies. Among them, PH type 1 is the most prevalent and severe type, and early end-stage renal failure often occurs.</p><p><strong>Summary: </strong>This review summarizes PH through pathophysiology, genotype, clinical manifestation, diagnosis, and treatment options. And explore the characteristics of Chinese PH patients.</p><p><strong>Key messages: </strong>Diagnosis of this rare disease is based on clinical symptoms, urinary or blood oxalate concentrations, liver biopsy, and genetic testing. Currently, the main treatment is massive hydration, citrate inhibition of crystallization, dialysis, liver and kidney transplantation, and pyridoxine. Recently, RNA interference drugs have also been used. In addition, technologies such as gene editing and autologous liver cell transplantation are also being developed. C.815_816insGA and c.33_34insC mutation in the <i>AGXT</i> gene could be a common variant in Chinese PH1 population. Mutations at the end of exon 6 account for approximately 50% of all Chinese HOGA1 mutations. Currently, the treatment of PH in China still relies mainly on symptomatic and high-throughput dialysis, with poor prognosis (especially for PH1 patients).</p>","PeriodicalId":17830,"journal":{"name":"Kidney Diseases","volume":"10 4","pages":"313-326"},"PeriodicalIF":3.1000,"publicationDate":"2024-06-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11309763/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Kidney Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1159/000539516","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
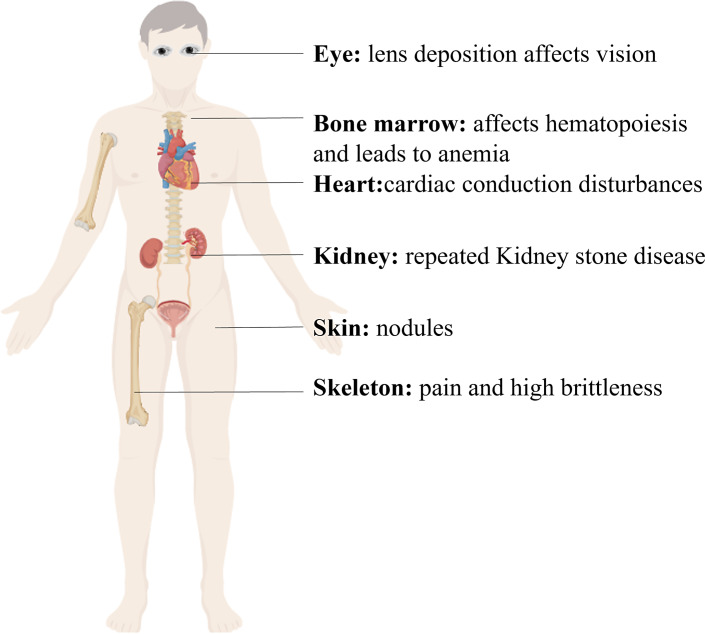
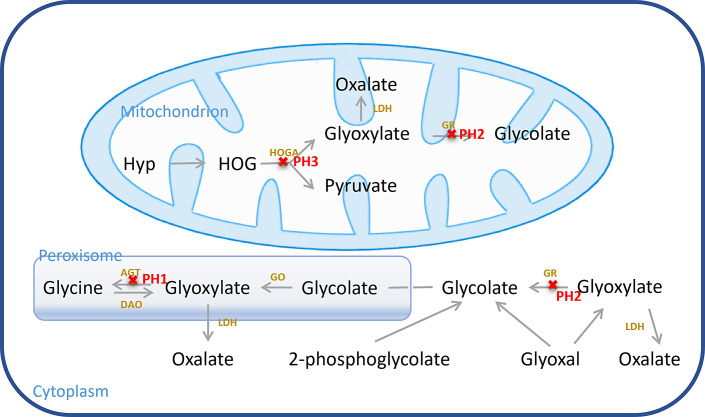
Background: Primary hyperoxaluria (PH) is a rare autosomal recessive disorder, mainly due to the increase in endogenous oxalate production, causing a series of clinical features such as kidney stones, nephrocalcinosis, progressive impairment of renal function, and systemic oxalosis. There are three common genetic causes of glycolate metabolism anomalies. Among them, PH type 1 is the most prevalent and severe type, and early end-stage renal failure often occurs.
Summary: This review summarizes PH through pathophysiology, genotype, clinical manifestation, diagnosis, and treatment options. And explore the characteristics of Chinese PH patients.
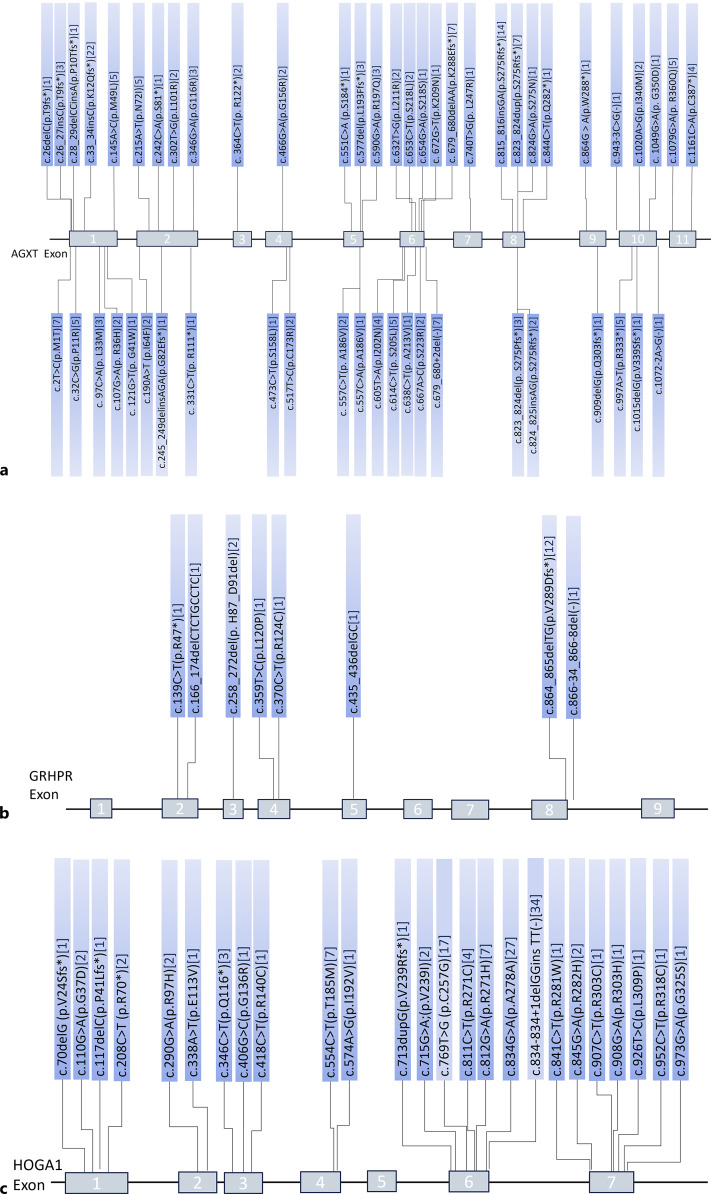
Key messages: Diagnosis of this rare disease is based on clinical symptoms, urinary or blood oxalate concentrations, liver biopsy, and genetic testing. Currently, the main treatment is massive hydration, citrate inhibition of crystallization, dialysis, liver and kidney transplantation, and pyridoxine. Recently, RNA interference drugs have also been used. In addition, technologies such as gene editing and autologous liver cell transplantation are also being developed. C.815_816insGA and c.33_34insC mutation in the AGXT gene could be a common variant in Chinese PH1 population. Mutations at the end of exon 6 account for approximately 50% of all Chinese HOGA1 mutations. Currently, the treatment of PH in China still relies mainly on symptomatic and high-throughput dialysis, with poor prognosis (especially for PH1 patients).
期刊介绍:
''Kidney Diseases'' aims to provide a platform for Asian and Western research to further and support communication and exchange of knowledge. Review articles cover the most recent clinical and basic science relevant to the entire field of nephrological disorders, including glomerular diseases, acute and chronic kidney injury, tubulo-interstitial disease, hypertension and metabolism-related disorders, end-stage renal disease, and genetic kidney disease. Special articles are prepared by two authors, one from East and one from West, which compare genetics, epidemiology, diagnosis methods, and treatment options of a disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


