A homozygous nonsense variant in the alternatively spliced VLDLR exon 4 causes a neurodevelopmental disorder without features of VLDLR cerebellar hypoplasia
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
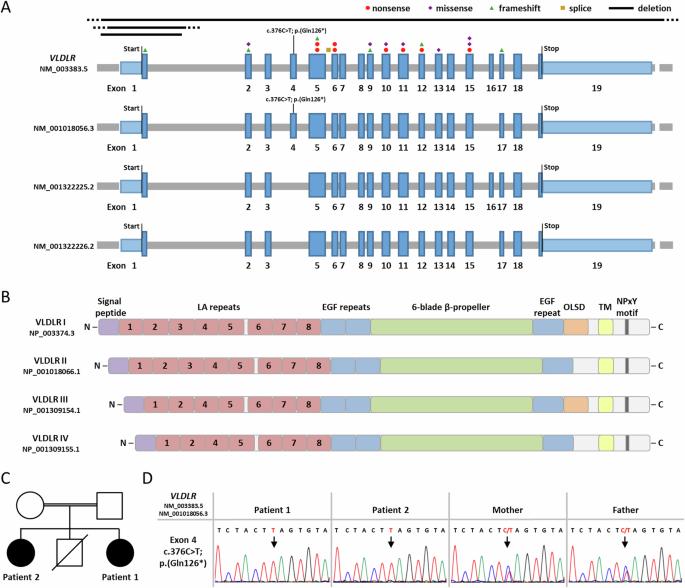
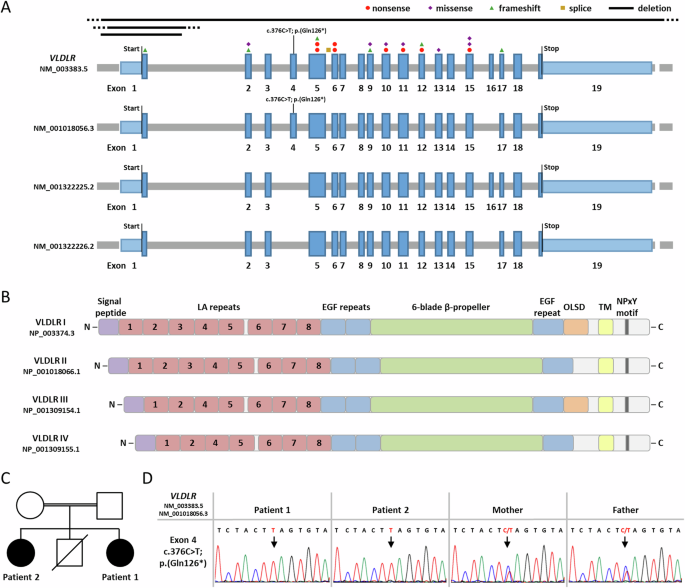
VLDLR cerebellar hypoplasia is characterized by intellectual disability, non-progressive cerebellar ataxia, and seizures. The characteristic MRI findings include hypoplasia of the inferior portion of the cerebellar vermis and hemispheres, simplified cortical gyration, and a small brain stem. Biallelic VLDLR pathogenic variants cause loss-of-function of the encoded very low-density lipoprotein receptor. VLDLR exons 4 and 16 are alternatively spliced, resulting in the expression of four transcript variants, including two exon 4-lacking mRNAs expressed in the human brain. Previously reported VLDLR pathogenic variants affect all four transcript variants. Here we report on two sisters with facial dysmorphism, microcephaly, intellectual disability, and normal brain imaging. Exome sequencing in one patient identified the homozygous VLDLR nonsense variant c.376C>T; p.(Gln126*) in exon 4; her similarly affected sister also carried the homozygous variant and parents were heterozygous carriers. VLDLR transcript analysis identified mRNAs with and without exon 4 in patient fibroblasts, while exon 4-containing VLDLR mRNAs were predominantly detected in control fibroblasts. We found significantly reduced VLDLR mRNA levels in patient compared to control cells, likely caused by nonsense-mediated mRNA decay of exon 4-containing VLDLR transcripts. Expression of neuronal VLDLR isoforms produced from exon 4-lacking transcripts may have protected both patients from developing the cerebellar hypoplasia phenotype.
交替剪接的 VLDLR 第 4 外显子中的一个同卵无义变体会导致一种神经发育障碍,但没有 VLDLR 小脑发育不全的特征。
VLDLR小脑发育不全症的特征是智力障碍、非进行性小脑共济失调和癫痫发作。其特征性核磁共振成像结果包括小脑蚓部和半球下部发育不良、皮质回旋简化和脑干较小。双叶 VLDLR 致病变体会导致编码的极低密度脂蛋白受体功能缺失。VLDLR 外显子 4 和 16 是交替剪接的,导致四种转录本变体的表达,包括在人脑中表达的两种缺乏外显子 4 的 mRNA。之前报道的 VLDLR 致病变体会影响所有四个转录本变体。在此,我们报告了两姐妹面部畸形、小头畸形、智力障碍和正常脑成像的情况。其中一名患者的外显子组测序确定了第 4 外显子中的同源 VLDLR 无义变异 c.376C>T;p. (Gln126*);她同样受影响的姐姐也携带同源变异,父母则是杂合携带者。VLDLR 转录本分析在患者成纤维细胞中发现了含或不含第 4 号外显子的 mRNA,而在对照组成纤维细胞中主要检测到含第 4 号外显子的 VLDLR mRNA。我们发现,与对照组细胞相比,患者细胞中的 VLDLR mRNA 水平明显降低,这可能是由于含外显子 4 的 VLDLR 转录物在无义介导的 mRNA 衰减所致。由缺乏外显子4的转录本产生的神经元VLDLR异构体的表达可能保护了这两名患者免于发展成小脑发育不全表型。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


