Mapping protein binding sites by photoreactive fragment pharmacophores
IF 5.9
2区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
Fragment screening is a popular strategy of generating viable chemical starting points especially for challenging targets. Although fragments provide a better coverage of chemical space and they have typically higher chance of binding, their weak affinity necessitates highly sensitive biophysical assays. Here, we introduce a screening concept that combines evolutionary optimized fragment pharmacophores with the use of a photoaffinity handle that enables high hit rates by LC-MS-based detection. The sensitivity of our screening protocol was further improved by a target-conjugated photocatalyst. We have designed, synthesized, and screened 100 diazirine-tagged fragments against three benchmark and three therapeutically relevant protein targets of different tractability. Our therapeutic targets included a conventional enzyme, the first bromodomain of BRD4, a protein-protein interaction represented by the oncogenic KRasG12D protein, and the yet unliganded N-terminal domain of the STAT5B transcription factor. We have discovered several fragment hits against all three targets and identified their binding sites via enzymatic digestion, structural studies and modeling. Our results revealed that this protocol outperforms screening traditional fully functionalized and photoaffinity fragments in better exploration of the available binding sites and higher hit rates observed for even difficult targets. Fragment screening is a popular strategy for generating viable chemical starting points for drug targets, however, weak affinity to targets, as well as the exploration of the binding site, remain challenging. Here, the authors develop pharmacophore-optimized photoaffinity fragments that can effectively explore the available binding site and enable a high hit rate and better sensitivity.
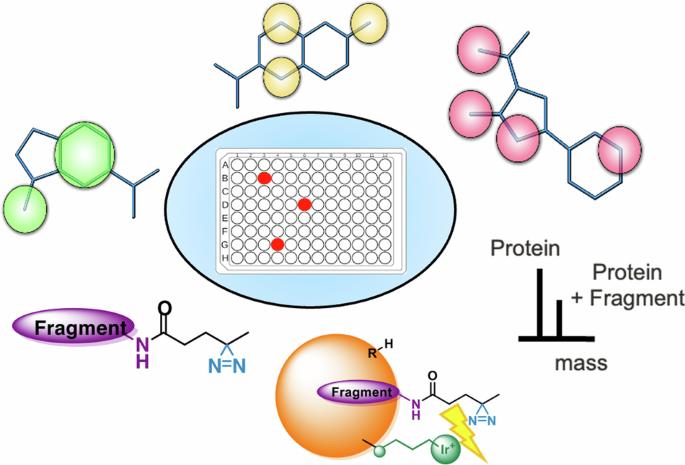
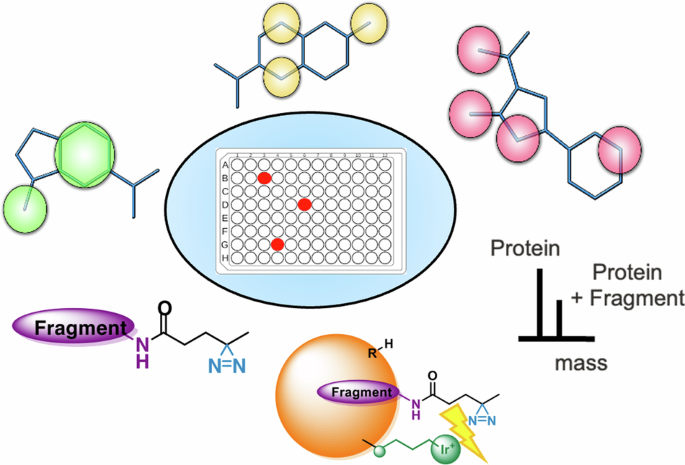
用光活性片段药剂绘制蛋白质结合位点图。
片段筛选是产生可行化学起点的一种常用策略,尤其适用于具有挑战性的靶标。虽然片段能更好地覆盖化学空间,而且它们通常有更高的结合几率,但由于它们的亲和力较弱,因此必须进行高灵敏度的生物物理检测。在这里,我们介绍了一种筛选概念,它将进化优化的片段药理作用与光亲和处理相结合,通过基于 LC-MS 的检测实现了高命中率。目标物结合的光催化剂进一步提高了筛选方案的灵敏度。我们设计、合成并筛选了 100 个重氮标记片段,它们分别针对三个基准蛋白质靶点和三个治疗相关蛋白质靶点。我们的治疗靶点包括一种传统酶、BRD4 的第一个溴结构域、以致癌 KRasG12D 蛋白为代表的蛋白间相互作用,以及 STAT5B 转录因子尚未连接的 N 端结构域。我们发现了针对这三个靶点的几个片段,并通过酶消化、结构研究和建模确定了它们的结合位点。我们的研究结果表明,在更好地探索可用的结合位点方面,该方案优于筛选传统的全功能化片段和光亲和片段,而且即使对困难的靶点也能观察到更高的命中率。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Communications Chemistry
Chemistry-General Chemistry
CiteScore
7.70
自引率
1.70%
发文量
146
审稿时长
13 weeks
期刊介绍:
Communications Chemistry is an open access journal from Nature Research publishing high-quality research, reviews and commentary in all areas of the chemical sciences. Research papers published by the journal represent significant advances bringing new chemical insight to a specialized area of research. We also aim to provide a community forum for issues of importance to all chemists, regardless of sub-discipline.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


