Efficient State-Specific Natural Orbital Based Equation of Motion Coupled Cluster Method for Core-Ionization Energies: Theory, Implementation, and Benchmark.
{"title":"Efficient State-Specific Natural Orbital Based Equation of Motion Coupled Cluster Method for Core-Ionization Energies: Theory, Implementation, and Benchmark.","authors":"Amrita Manna, Bhavnesh Jangid, Rakesh Pant, Achintya Kumar Dutta","doi":"10.1021/acs.jctc.4c00546","DOIUrl":null,"url":null,"abstract":"<p><p>We have implemented a reduced-cost partial triples correction scheme to the equation of motion coupled cluster method for core-ionization energy based on state-specific natural orbitals. The second-order Algebraic Diagrammatic Construction (ADC) method is used to generate the state-specific natural orbital, which provides quicker convergence of the core-IP value with respect to the size of the virtual space than that observed in standard MP2-based natural orbitals. The error due to truncation of the virtual orbital can be reduced by using a perturbative correction. The accuracy of the method can be controlled by a single threshold, and there is a black box to use. The inclusion of the partial triples correction in the natural orbital based EOM-CCSD method greatly improves the agreement of the results with the experiment. The efficiency of the present implementation is demonstrated by calculating the core-ionization energy of a molecule containing 60 atoms and more than 2000 basis functions.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00546","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/29 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
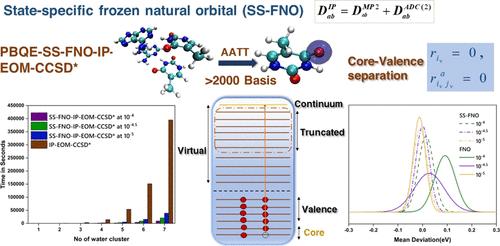
We have implemented a reduced-cost partial triples correction scheme to the equation of motion coupled cluster method for core-ionization energy based on state-specific natural orbitals. The second-order Algebraic Diagrammatic Construction (ADC) method is used to generate the state-specific natural orbital, which provides quicker convergence of the core-IP value with respect to the size of the virtual space than that observed in standard MP2-based natural orbitals. The error due to truncation of the virtual orbital can be reduced by using a perturbative correction. The accuracy of the method can be controlled by a single threshold, and there is a black box to use. The inclusion of the partial triples correction in the natural orbital based EOM-CCSD method greatly improves the agreement of the results with the experiment. The efficiency of the present implementation is demonstrated by calculating the core-ionization energy of a molecule containing 60 atoms and more than 2000 basis functions.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


