Sintering Kinetics and Interfacial Heat Transfer Process of Binary Alloy Nanoparticles Catalysts: Molecular Dynamics Simulation
IF 3.8
3区 工程技术
Q2 ENGINEERING, CHEMICAL
引用次数: 0
Abstract
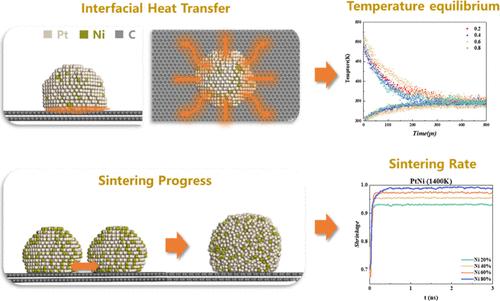
The thermal stability of supported alloy nanocatalysts is a crucial factor limiting the lifespan of catalysts. In this study, molecular dynamics (MD) simulations were employed to investigate the interface heat transfer and sintering kinetics of binary alloy nanoparticles composed of Pt, Ni, and Fe supported on graphene substrates. Analysis of the crystalline distribution, radial distribution function (RDF), and potential energy variations of the supported particles revealed that significant differences in the atomic radii and interaction energies (potentials) of the metals could lead to the formation of core–shell structures in alloy nanoparticles, whereas the reverse scenario might result in disordered alloy structures, with the particle structure closely tied to its thermal stability. Simulation results of the heat transfer process indicated that core–shell structures could enhance the cooling rate of the particles. Additionally, metal with superior thermal conductivity could increase the contact area between the particles and the substrate, thereby enhancing the interfacial thermal conductivity. Further analysis of the shrinkage of each alloy revealed that when a more stable (judged by the maximum shrinkage of the particles within the same time) metal comprised a higher proportion of the alloy, the particles exhibited greater stability and were less prone to sintering.
二元合金纳米颗粒催化剂的烧结动力学和界面传热过程:分子动力学模拟
支撑合金纳米催化剂的热稳定性是限制催化剂寿命的关键因素。本研究采用分子动力学(MD)模拟研究了石墨烯基底上支撑的由铂、镍和铁组成的二元合金纳米颗粒的界面传热和烧结动力学。对支撑颗粒的结晶分布、径向分布函数(RDF)和势能变化的分析表明,金属原子半径和相互作用能(势能)的显著差异可能导致合金纳米颗粒形成核壳结构,而相反的情况则可能导致无序合金结构,颗粒结构与其热稳定性密切相关。传热过程的模拟结果表明,核壳结构可以提高颗粒的冷却速度。此外,导热性能优异的金属可增加颗粒与基底之间的接触面积,从而提高界面导热率。对每种合金收缩率的进一步分析表明,当更稳定(以颗粒在相同时间内的最大收缩率来判断)的金属在合金中所占比例较高时,颗粒表现出更高的稳定性,不易烧结。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Industrial & Engineering Chemistry Research
工程技术-工程:化工
CiteScore
7.40
自引率
7.10%
发文量
1467
审稿时长
2.8 months
期刊介绍:
ndustrial & Engineering Chemistry, with variations in title and format, has been published since 1909 by the American Chemical Society. Industrial & Engineering Chemistry Research is a weekly publication that reports industrial and academic research in the broad fields of applied chemistry and chemical engineering with special focus on fundamentals, processes, and products.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


