A metagenomic next-generation sequencing (mNGS)-based analysis of bronchoalveolar lavage samples in patients with an acute exacerbation of chronic obstructive pulmonary disease
Junfang Wu, Yongqing Zhang, Jinjin Duan, Yiqun Wei, Yi Miao
{"title":"A metagenomic next-generation sequencing (mNGS)-based analysis of bronchoalveolar lavage samples in patients with an acute exacerbation of chronic obstructive pulmonary disease","authors":"Junfang Wu, Yongqing Zhang, Jinjin Duan, Yiqun Wei, Yi Miao","doi":"10.1007/s10735-024-10225-1","DOIUrl":null,"url":null,"abstract":"<div><p>The role of the bronchoalveolar lavage fluid (BALF) microbiome in acute exacerbations of chronic obstructive pulmonary disease (AECOPD) remains unclear. The advent of the metagenomic next-generation sequencing (mNGS) has made it possible to reveal the complex microbiome composition of the respiratory tract. This study aimed to explore whether there are differences in the BALF microbiome of AECOPD patients with different lung functions. We enrolled 55 AECOPD patients and divided them into a mild group (n = 31) and a severe group (n = 24) according to their lung function. We collected BALF and submitted it to mNGS and bioinformatics analysis. At the species level, mNGS identified 264 bacteria, 13 fungi and 12 viruses in the mild group, and 174 bacteria, 6 fungi and 6 viruses in the severe group. Mixed bacterial and viral infection occurred in both groups. At the genus level, <i>Rothia</i> and <i>Veillonella</i> were more abundant in the mild group, while <i>Pseudomonas</i> and <i>Staphylococcus</i> were more abundant in the severe group. At the species level, compared with the mild group, the relative abundance of <i>Haemophilus influenzae</i> and <i>Pseudomonas aeruginosa</i> was increased in the severe group. Besides, the BALF microbiome composition was similar between the two groups, and there was no significant difference in α and β diversity. Forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC) (%) showed no significant correlation with the Shannon or Simpson index. The microbiome abundance was different between the mild and severe groups; however, microbiome diversity was similar between the two groups. Based on our findings, <i>Haemophilus influenzae</i> and <i>Pseudomonas aeruginosa</i> may be the pathogenic bacteria that cause the difference in lung function in patients with AECOPD.</p></div>","PeriodicalId":650,"journal":{"name":"Journal of Molecular Histology","volume":"55 5","pages":"709 - 719"},"PeriodicalIF":2.9000,"publicationDate":"2024-07-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Histology","FirstCategoryId":"99","ListUrlMain":"https://link.springer.com/article/10.1007/s10735-024-10225-1","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
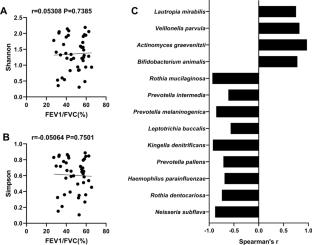
The role of the bronchoalveolar lavage fluid (BALF) microbiome in acute exacerbations of chronic obstructive pulmonary disease (AECOPD) remains unclear. The advent of the metagenomic next-generation sequencing (mNGS) has made it possible to reveal the complex microbiome composition of the respiratory tract. This study aimed to explore whether there are differences in the BALF microbiome of AECOPD patients with different lung functions. We enrolled 55 AECOPD patients and divided them into a mild group (n = 31) and a severe group (n = 24) according to their lung function. We collected BALF and submitted it to mNGS and bioinformatics analysis. At the species level, mNGS identified 264 bacteria, 13 fungi and 12 viruses in the mild group, and 174 bacteria, 6 fungi and 6 viruses in the severe group. Mixed bacterial and viral infection occurred in both groups. At the genus level, Rothia and Veillonella were more abundant in the mild group, while Pseudomonas and Staphylococcus were more abundant in the severe group. At the species level, compared with the mild group, the relative abundance of Haemophilus influenzae and Pseudomonas aeruginosa was increased in the severe group. Besides, the BALF microbiome composition was similar between the two groups, and there was no significant difference in α and β diversity. Forced expiratory volume in 1 s/forced vital capacity (FEV1/FVC) (%) showed no significant correlation with the Shannon or Simpson index. The microbiome abundance was different between the mild and severe groups; however, microbiome diversity was similar between the two groups. Based on our findings, Haemophilus influenzae and Pseudomonas aeruginosa may be the pathogenic bacteria that cause the difference in lung function in patients with AECOPD.
期刊介绍:
The Journal of Molecular Histology publishes results of original research on the localization and expression of molecules in animal cells, tissues and organs. Coverage includes studies describing novel cellular or ultrastructural distributions of molecules which provide insight into biochemical or physiological function, development, histologic structure and disease processes.
Major research themes of particular interest include:
- Cell-Cell and Cell-Matrix Interactions;
- Connective Tissues;
- Development and Disease;
- Neuroscience.
Please note that the Journal of Molecular Histology does not consider manuscripts dealing with the application of immunological or other probes on non-standard laboratory animal models unless the results are clearly of significant and general biological importance.
The Journal of Molecular Histology publishes full-length original research papers, review articles, short communications and letters to the editors. All manuscripts are typically reviewed by two independent referees. The Journal of Molecular Histology is a continuation of The Histochemical Journal.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


