{"title":"Arabidopsis enters the single-cell proteomics era","authors":"Monique van Schie, Dolf Weijers","doi":"10.1111/nph.19992","DOIUrl":null,"url":null,"abstract":"<p>Despite their biological relevance, proteins are difficult to identify and quantify, since it is impossible to amplify the signal of proteins in a way similar to PCR for DNA and RNA. Hence, measurements need to be able to detect minute quantities. Given that different proteins may accumulate at vastly different levels in cells, methods also need to accommodate signals that range up to seven orders of magnitude. Modern mass spectrometers used for proteomics are improving rapidly, both in sensitivity and sample throughput. However, single-cell measurements still pose two major challenges. The first is in handling the samples and extracting the peptides. Commonly, cell types are selected by FACS sorting and processed using a pipetting robot in minute volumes, during which caution needs to be taken to limit pipetting steps to avoid loss of material (Petelski <i>et al</i>., <span>2021</span>; Sun & Kumar, <span>2022</span>; Matzinger <i>et al</i>., <span>2023</span>; Ctortecka <i>et al</i>., <span>2024</span>). In plants, there is an extra challenge in the form of the cell wall, which Montes <i>et al</i>. dealt with by FACS sorting protoplasts after cell walls had been enzymatically removed (Fig. 1). Furthermore, plants make more metabolites compared to animals, and such compounds can interfere with protein identification in mass spectrometry. To solve this, Montes <i>et al</i>. used an optimized sample cleanup protocol (Song <i>et al</i>., <span>2018</span>). Lastly, the spectra produced by mass spectrometers need to be interpreted, and the algorithms for which are under continuous development. There are various approaches under active development, varying in acquisition methods and the use or omission of isotopic labels (Truong & Kelly, <span>2024</span>). The most suitable approach depends on the study requirements and available resources but is critical for the outcome. To summarize, every step of a single-cell proteomics approach needs to be optimized and performed almost perfectly to obtain useful results (Fig. 1).</p><p>Despite the challenging aspects, Montes <i>et al</i>. were able to identify and quantify proteins in 81 cells that passed quality control filters. Collectively, this allowed them to identify and quantify 3217 proteins across two cell types from Arabidopsis roots, endodermis and cortex. Among those proteins, 596 were significantly enriched in one or the other cell type. In general, the highest expressing proteins are measured when using mass spectrometry, which are often ‘housekeeping’ or structural proteins. Proteins such as transcription factors can be expressed at levels as low as a single copy per cell. Montes <i>et al</i>. identified 188 transcription factors and 175 kinases in their total dataset, which suggests that the method allows the collection of information at a depth compatible with studying regulatory events in cell biology. The authors furthermore show they identify a clear proteomic signature that separates the cortex and endodermis cells, which are physically neighboring cells. However, interpretation of cell-level data should be carried out with caution since cell size can influence the observed ratios of proteins in single-cell experiments. It is important to ensure that variation in protein quantifications is a feature of the biology of the cell and not a technical artefact (Lanz <i>et al</i>., <span>2023</span>). The effect of cell size variation can be filtered out during data analysis as done by Montes <i>et al</i>., but a future improvement could be to <i>a priori</i> select cells within a specific size range.</p><p>The data presented by Montes <i>et al</i>. should be seen as a promising proof of concept, which opens the door to many interesting experiments in the future. Single-cell proteomics is rapidly gaining popularity and is improving in depth, quality, and throughput. Roughly a year ago, 5000 proteins identified per experiment was deemed to be a good goal to strive for when working on biologically relevant questions (Rosenberger <i>et al</i>., <span>2023</span>), but this number has been surpassed already (Truong & Kelly, <span>2024</span>). One future point of investigation could be constructing cell-specific protein expression atlases comparable to the current single-cell RNA-Seq atlases. Furthermore, the proteome of highly specialized cells could be measured during their developmental trajectory. This could give relevant insights into their differentiation and functioning and help identify the key proteins acting in these processes. Single-cell proteomics could also be leveraged to obtain the spatial resolution of certain processes, such as cell-type-specific responses to invading pathogens or abiotic stresses. In the future, it might even be possible to add analysis of posttranslational modifications, proteoforms and metabolites to obtain even deeper biological insights. Advanced methods to analyze all these data will also be necessary, for example, to combine single-cell RNA-Seq and single-cell proteomics data to gain insight of the genetic regulation of protein content at the single-cell level (Xu <i>et al</i>., <span>2024</span>). The amount of data currently produced is beyond the comprehension of the human brain, so caution should be taken when experimental designs and data analysis pipelines are created, to be able to extract meaningful insights from the data.</p><p>In general, the advances required to make single-cell proteomics possible will also trickle down to the wider proteomics field, for example, to reduce input material while gaining depth on bulk experiments and increase throughput and resolution on techniques such as imaging mass spectrometry (Su <i>et al</i>., <span>2024</span>). When the amount of input material can be reduced and the number of measured samples per day increased, it will be possible to realize radically different and larger-scale study designs. When doing treatments, for example, multiple time points and concentrations could be compared, or processes such as differentiation of tissues and the response to stresses can be studied with higher time resolution. Furthermore, techniques that do not require cell wall removal such as deep visual proteomics, where the proteins from tissue sections are directly measured with cellular resolution, could be applied (Mund <i>et al</i>., <span>2022</span>; Su <i>et al</i>., <span>2024</span>). Generating protoplasts from tissues will likely alter their proteome. As mentioned by Montes <i>et al</i>., it is very important that the effect of cell wall digestion on plant cells is characterized, for example by comparing the proteomic bulk data of tissues and the corresponding protoplasts.</p><p>In conclusion, it is safe to say that Arabidopsis has successfully entered the single-cell proteomics era and many exciting discoveries are waiting in this rapidly growing and developing field. Time will tell, but getting a complete picture of the differences between one cell and its neighbor, and how these differences contribute to the phenotype, does not seem that far-fetched anymore.</p>","PeriodicalId":214,"journal":{"name":"New Phytologist","volume":"244 5","pages":"1678-1680"},"PeriodicalIF":8.3000,"publicationDate":"2024-07-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/nph.19992","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Phytologist","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/nph.19992","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PLANT SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
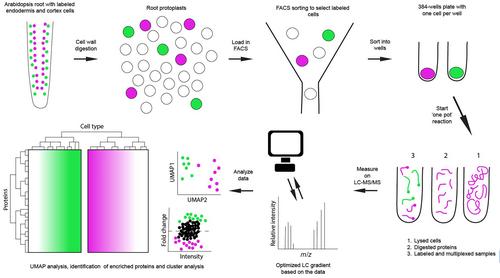
Despite their biological relevance, proteins are difficult to identify and quantify, since it is impossible to amplify the signal of proteins in a way similar to PCR for DNA and RNA. Hence, measurements need to be able to detect minute quantities. Given that different proteins may accumulate at vastly different levels in cells, methods also need to accommodate signals that range up to seven orders of magnitude. Modern mass spectrometers used for proteomics are improving rapidly, both in sensitivity and sample throughput. However, single-cell measurements still pose two major challenges. The first is in handling the samples and extracting the peptides. Commonly, cell types are selected by FACS sorting and processed using a pipetting robot in minute volumes, during which caution needs to be taken to limit pipetting steps to avoid loss of material (Petelski et al., 2021; Sun & Kumar, 2022; Matzinger et al., 2023; Ctortecka et al., 2024). In plants, there is an extra challenge in the form of the cell wall, which Montes et al. dealt with by FACS sorting protoplasts after cell walls had been enzymatically removed (Fig. 1). Furthermore, plants make more metabolites compared to animals, and such compounds can interfere with protein identification in mass spectrometry. To solve this, Montes et al. used an optimized sample cleanup protocol (Song et al., 2018). Lastly, the spectra produced by mass spectrometers need to be interpreted, and the algorithms for which are under continuous development. There are various approaches under active development, varying in acquisition methods and the use or omission of isotopic labels (Truong & Kelly, 2024). The most suitable approach depends on the study requirements and available resources but is critical for the outcome. To summarize, every step of a single-cell proteomics approach needs to be optimized and performed almost perfectly to obtain useful results (Fig. 1).
Despite the challenging aspects, Montes et al. were able to identify and quantify proteins in 81 cells that passed quality control filters. Collectively, this allowed them to identify and quantify 3217 proteins across two cell types from Arabidopsis roots, endodermis and cortex. Among those proteins, 596 were significantly enriched in one or the other cell type. In general, the highest expressing proteins are measured when using mass spectrometry, which are often ‘housekeeping’ or structural proteins. Proteins such as transcription factors can be expressed at levels as low as a single copy per cell. Montes et al. identified 188 transcription factors and 175 kinases in their total dataset, which suggests that the method allows the collection of information at a depth compatible with studying regulatory events in cell biology. The authors furthermore show they identify a clear proteomic signature that separates the cortex and endodermis cells, which are physically neighboring cells. However, interpretation of cell-level data should be carried out with caution since cell size can influence the observed ratios of proteins in single-cell experiments. It is important to ensure that variation in protein quantifications is a feature of the biology of the cell and not a technical artefact (Lanz et al., 2023). The effect of cell size variation can be filtered out during data analysis as done by Montes et al., but a future improvement could be to a priori select cells within a specific size range.
The data presented by Montes et al. should be seen as a promising proof of concept, which opens the door to many interesting experiments in the future. Single-cell proteomics is rapidly gaining popularity and is improving in depth, quality, and throughput. Roughly a year ago, 5000 proteins identified per experiment was deemed to be a good goal to strive for when working on biologically relevant questions (Rosenberger et al., 2023), but this number has been surpassed already (Truong & Kelly, 2024). One future point of investigation could be constructing cell-specific protein expression atlases comparable to the current single-cell RNA-Seq atlases. Furthermore, the proteome of highly specialized cells could be measured during their developmental trajectory. This could give relevant insights into their differentiation and functioning and help identify the key proteins acting in these processes. Single-cell proteomics could also be leveraged to obtain the spatial resolution of certain processes, such as cell-type-specific responses to invading pathogens or abiotic stresses. In the future, it might even be possible to add analysis of posttranslational modifications, proteoforms and metabolites to obtain even deeper biological insights. Advanced methods to analyze all these data will also be necessary, for example, to combine single-cell RNA-Seq and single-cell proteomics data to gain insight of the genetic regulation of protein content at the single-cell level (Xu et al., 2024). The amount of data currently produced is beyond the comprehension of the human brain, so caution should be taken when experimental designs and data analysis pipelines are created, to be able to extract meaningful insights from the data.
In general, the advances required to make single-cell proteomics possible will also trickle down to the wider proteomics field, for example, to reduce input material while gaining depth on bulk experiments and increase throughput and resolution on techniques such as imaging mass spectrometry (Su et al., 2024). When the amount of input material can be reduced and the number of measured samples per day increased, it will be possible to realize radically different and larger-scale study designs. When doing treatments, for example, multiple time points and concentrations could be compared, or processes such as differentiation of tissues and the response to stresses can be studied with higher time resolution. Furthermore, techniques that do not require cell wall removal such as deep visual proteomics, where the proteins from tissue sections are directly measured with cellular resolution, could be applied (Mund et al., 2022; Su et al., 2024). Generating protoplasts from tissues will likely alter their proteome. As mentioned by Montes et al., it is very important that the effect of cell wall digestion on plant cells is characterized, for example by comparing the proteomic bulk data of tissues and the corresponding protoplasts.
In conclusion, it is safe to say that Arabidopsis has successfully entered the single-cell proteomics era and many exciting discoveries are waiting in this rapidly growing and developing field. Time will tell, but getting a complete picture of the differences between one cell and its neighbor, and how these differences contribute to the phenotype, does not seem that far-fetched anymore.
期刊介绍:
New Phytologist is an international electronic journal published 24 times a year. It is owned by the New Phytologist Foundation, a non-profit-making charitable organization dedicated to promoting plant science. The journal publishes excellent, novel, rigorous, and timely research and scholarship in plant science and its applications. The articles cover topics in five sections: Physiology & Development, Environment, Interaction, Evolution, and Transformative Plant Biotechnology. These sections encompass intracellular processes, global environmental change, and encourage cross-disciplinary approaches. The journal recognizes the use of techniques from molecular and cell biology, functional genomics, modeling, and system-based approaches in plant science. Abstracting and Indexing Information for New Phytologist includes Academic Search, AgBiotech News & Information, Agroforestry Abstracts, Biochemistry & Biophysics Citation Index, Botanical Pesticides, CAB Abstracts®, Environment Index, Global Health, and Plant Breeding Abstracts, and others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


