Sources of gene expression variation in a globally diverse human cohort
IF 50.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
Genetic variation that influences gene expression and splicing is a key source of phenotypic diversity1–5. Although invaluable, studies investigating these links in humans have been strongly biased towards participants of European ancestries, which constrains generalizability and hinders evolutionary research. Here to address these limitations, we developed MAGE, an open-access RNA sequencing dataset of lymphoblastoid cell lines from 731 individuals from the 1000 Genomes Project6, spread across 5 continental groups and 26 populations. Most variation in gene expression (92%) and splicing (95%) was distributed within versus between populations, which mirrored the variation in DNA sequence. We mapped associations between genetic variants and expression and splicing of nearby genes (cis-expression quantitative trait loci (eQTLs) and cis-splicing QTLs (sQTLs), respectively). We identified more than 15,000 putatively causal eQTLs and more than 16,000 putatively causal sQTLs that are enriched for relevant epigenomic signatures. These include 1,310 eQTLs and 1,657 sQTLs that are largely private to underrepresented populations. Our data further indicate that the magnitude and direction of causal eQTL effects are highly consistent across populations. Moreover, the apparent ‘population-specific’ effects observed in previous studies were largely driven by low resolution or additional independent eQTLs of the same genes that were not detected. Together, our study expands our understanding of human gene expression diversity and provides an inclusive resource for studying the evolution and function of human genomes. A new open-access RNA sequencing dataset, MAGE, of 731 individuals across geographically diverse human populations provides a valuable resource to study genetic diversity and evolution and expands the capacity to identify new genetic associations.
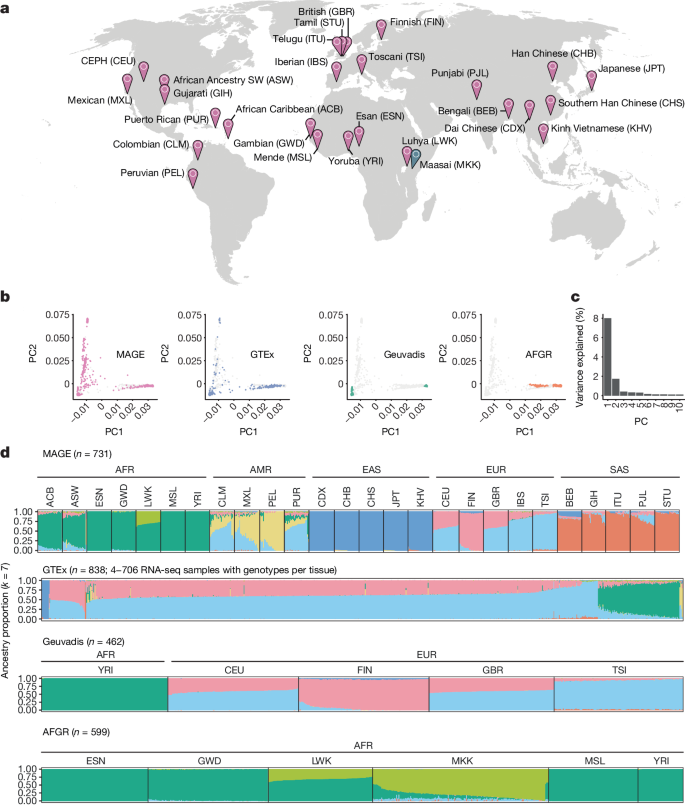
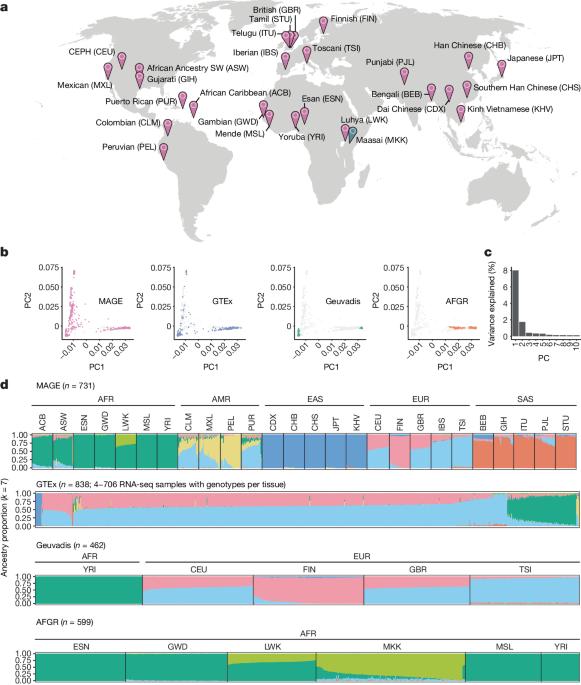
全球不同人群基因表达变异的来源。
影响基因表达和剪接的遗传变异是表型多样性的一个重要来源1-5。尽管这些研究很有价值,但调查人类中这些联系的研究却严重偏向于欧洲血统的参与者,这限制了研究的普遍性并阻碍了进化研究。为了解决这些局限性,我们开发了 MAGE,这是一个开放存取的 RNA 测序数据集,来自 1000 基因组计划6 的 731 个个体的淋巴母细胞系,分布在 5 个大陆组和 26 个人群中。基因表达(92%)和剪接(95%)的大多数变异分布在人群内部和人群之间,这反映了 DNA 序列的变异。我们绘制了遗传变异与附近基因的表达和剪接之间的关联图(分别为顺式表达数量性状位点(eQTLs)和顺式剪接 QTLs(sQTLs))。我们发现了超过 15,000 个推测为因果关系的 eQTL 和超过 16,000 个推测为因果关系的 sQTL,它们富集了相关的表观基因组特征。其中包括 1,310 个 eQTL 和 1,657 个 sQTL,它们在很大程度上不属于代表性不足的人群。我们的数据进一步表明,eQTL 的因果效应的大小和方向在不同人群中高度一致。此外,以往研究中观察到的明显的 "种群特异性 "效应在很大程度上是由低分辨率或未检测到的相同基因的额外独立 eQTLs 驱动的。总之,我们的研究拓展了我们对人类基因表达多样性的理解,为研究人类基因组的进化和功能提供了一个包容性资源。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
文献相关原料
| 公司名称 | 产品信息 | 采购帮参考价格 |
|---|
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


