Flavonoid as a Potent Antioxidant: Quantitative Structure-Activity Relationship Analysis, Mechanism Study, and Molecular Design by Synergizing Molecular Simulation and Machine Learning.
{"title":"Flavonoid as a Potent Antioxidant: Quantitative Structure-Activity Relationship Analysis, Mechanism Study, and Molecular Design by Synergizing Molecular Simulation and Machine Learning.","authors":"Ling Lu, Yajie Luan, Huaqi Wang, Yangyang Gao, Sizhu Wu, Xiuying Zhao","doi":"10.1021/acs.jpca.4c03241","DOIUrl":null,"url":null,"abstract":"<p><p>In this work, a quantitative structure-antioxidant activity relationship of flavonoids was performed using a machine learning (ML) method. To achieve lipid-soluble, highly antioxidant flavonoids, 398 molecular structures with various substitute groups were designed based on the flavonoid skeleton. The hydrogen dissociation energies (Δ<i>G</i><sub>1</sub>, Δ<i>G</i><sub>2</sub>, and Δ<i>G</i><sub>3</sub>) related to multiple hydrogen atom transfer processes and the solubility parameter (δ) of flavonoids were calculated using molecular simulation. The group decomposition results and the calculated antioxidant parameters constituted the ML data set. The artificial neural network and random forest models were constructed to predict and analyze the contribution of the substitute groups and positions to the antioxidant activity. The results showed the hydroxyl group at positions B4', B5', and B6' and the branched alkyl group at position C3 in the flavonoid skeleton were the optimal choice for improving antioxidant activity and compatibility with apolar organic materials. Compared to the pyrogallol group-grafted flavonoid, the designed potent flavonoid decreased Δ<i>G</i><sub>1</sub> and δ by 2.2 and 15.1%, respectively, while Δ<i>G</i><sub>2</sub> and Δ<i>G</i><sub>3</sub> kept the favorable lower values. These findings suggest that an efficient flavonoid prefers multiple ortho-phenolic hydroxyl groups and suitable sites with hydrophobic groups. The combination of molecular simulation and the ML method may offer a new research approach for the molecular design of novel antioxidants.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c03241","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/18 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
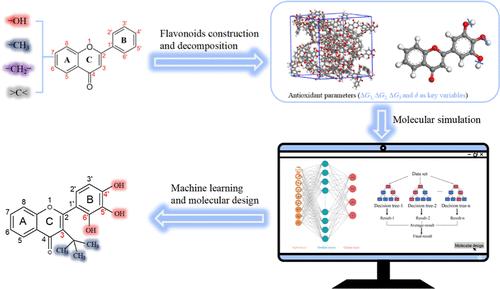
In this work, a quantitative structure-antioxidant activity relationship of flavonoids was performed using a machine learning (ML) method. To achieve lipid-soluble, highly antioxidant flavonoids, 398 molecular structures with various substitute groups were designed based on the flavonoid skeleton. The hydrogen dissociation energies (ΔG1, ΔG2, and ΔG3) related to multiple hydrogen atom transfer processes and the solubility parameter (δ) of flavonoids were calculated using molecular simulation. The group decomposition results and the calculated antioxidant parameters constituted the ML data set. The artificial neural network and random forest models were constructed to predict and analyze the contribution of the substitute groups and positions to the antioxidant activity. The results showed the hydroxyl group at positions B4', B5', and B6' and the branched alkyl group at position C3 in the flavonoid skeleton were the optimal choice for improving antioxidant activity and compatibility with apolar organic materials. Compared to the pyrogallol group-grafted flavonoid, the designed potent flavonoid decreased ΔG1 and δ by 2.2 and 15.1%, respectively, while ΔG2 and ΔG3 kept the favorable lower values. These findings suggest that an efficient flavonoid prefers multiple ortho-phenolic hydroxyl groups and suitable sites with hydrophobic groups. The combination of molecular simulation and the ML method may offer a new research approach for the molecular design of novel antioxidants.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


