Olivia Castellini-Pérez, Elena Povedano, Guillermo Barturen, Manuel Martínez-Bueno, Andrii Iakovliev, Martin Kerick, Raúl López-Domínguez, Concepción Marañón, Javier Martín, Esteban Ballestar, María Orietta Borghi, Weiliang Qiu, Cheng Zhu, Srinivas Shankara, Athina Spiliopoulou, Emanuele de Rinaldis, Elena Carnero-Montoro, Marta E Alarcón-Riquelme
{"title":"Molecular subtypes explain lupus epigenomic heterogeneity unveiling new regulatory genetic risk variants.","authors":"Olivia Castellini-Pérez, Elena Povedano, Guillermo Barturen, Manuel Martínez-Bueno, Andrii Iakovliev, Martin Kerick, Raúl López-Domínguez, Concepción Marañón, Javier Martín, Esteban Ballestar, María Orietta Borghi, Weiliang Qiu, Cheng Zhu, Srinivas Shankara, Athina Spiliopoulou, Emanuele de Rinaldis, Elena Carnero-Montoro, Marta E Alarcón-Riquelme","doi":"10.1038/s41525-024-00420-0","DOIUrl":null,"url":null,"abstract":"<p><p>The heterogeneity of systemic lupus erythematosus (SLE) can be explained by epigenetic alterations that disrupt transcriptional programs mediating environmental and genetic risk. This study evaluated the epigenetic contribution to SLE heterogeneity considering molecular and serological subtypes, genetics and transcriptional status, followed by drug target discovery. We performed a stratified epigenome-wide association studies of whole blood DNA methylation from 213 SLE patients and 221 controls. Methylation quantitative trait loci analyses, cytokine and transcription factor activity - epigenetic associations and methylation-expression correlations were conducted. New drug targets were searched for based on differentially methylated genes. In a stratified approach, a total of 974 differential methylation CpG sites with dependency on molecular subtypes and autoantibody profiles were found. Mediation analyses suggested that SLE-associated SNPs in the HLA region exert their risk through DNA methylation changes. Novel genetic variants regulating DNAm in disease or in specific molecular contexts were identified. The epigenetic landscapes showed strong association with transcription factor activity and cytokine levels, conditioned by the molecular context. Epigenetic signals were enriched in known and novel drug targets for SLE. This study reveals possible genetic drivers and consequences of epigenetic variability on SLE heterogeneity and disentangles the DNAm mediation role on SLE genetic risk and novel disease-specific meQTLs. Finally, novel targets for drug development were discovered.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"38"},"PeriodicalIF":4.8000,"publicationDate":"2024-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11252280/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-024-00420-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
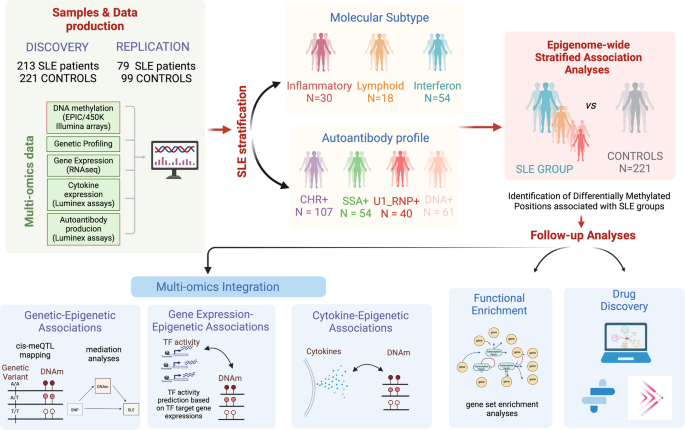
The heterogeneity of systemic lupus erythematosus (SLE) can be explained by epigenetic alterations that disrupt transcriptional programs mediating environmental and genetic risk. This study evaluated the epigenetic contribution to SLE heterogeneity considering molecular and serological subtypes, genetics and transcriptional status, followed by drug target discovery. We performed a stratified epigenome-wide association studies of whole blood DNA methylation from 213 SLE patients and 221 controls. Methylation quantitative trait loci analyses, cytokine and transcription factor activity - epigenetic associations and methylation-expression correlations were conducted. New drug targets were searched for based on differentially methylated genes. In a stratified approach, a total of 974 differential methylation CpG sites with dependency on molecular subtypes and autoantibody profiles were found. Mediation analyses suggested that SLE-associated SNPs in the HLA region exert their risk through DNA methylation changes. Novel genetic variants regulating DNAm in disease or in specific molecular contexts were identified. The epigenetic landscapes showed strong association with transcription factor activity and cytokine levels, conditioned by the molecular context. Epigenetic signals were enriched in known and novel drug targets for SLE. This study reveals possible genetic drivers and consequences of epigenetic variability on SLE heterogeneity and disentangles the DNAm mediation role on SLE genetic risk and novel disease-specific meQTLs. Finally, novel targets for drug development were discovered.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


