Heterozygous mutations in the straitjacket region of the latency-associated peptide domain of TGFB2 cause Camurati–Engelmann disease type II
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
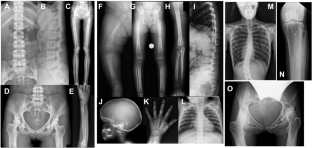
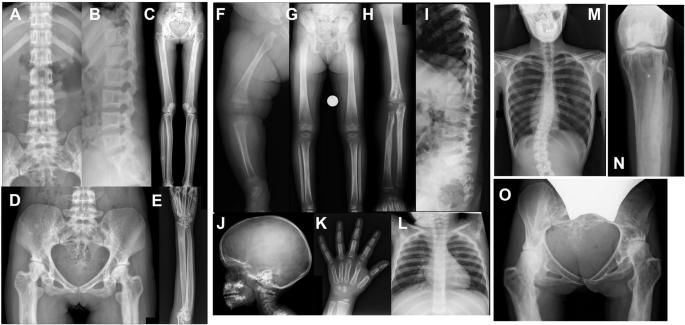
Camurati–Engelmann disease (CED) is an autosomal dominant bone dysplasia characterized by progressive hyperostosis of the skull base and diaphyses of the long bones. CED is further divided into two subtypes, CED1 and CED2, according to the presence or absence of TGFB1 mutations, respectively. In this study, we used exome sequencing to investigate the genetic cause of CED2 in three pedigrees and identified two de novo heterozygous mutations in TGFB2 among the three patients. Both mutations were located in the region of the gene encoding the straitjacket subdomain of the latency-associated peptide (LAP) of pro-TGF-β2. Structural simulations of the mutant LAPs suggested that the mutations could cause significant conformational changes and lead to a reduction in TGF-β2 inactivation. An activity assay confirmed a significant increase in TGF-β2/SMAD signaling. In vitro osteogenic differentiation experiment using iPS cells from one of the CED2 patients showed significantly enhanced ossification, suggesting that the pathogenic mechanism of CED2 is increased activation of TGF-β2 by loss-of-function of the LAP. These results, in combination with the difference in hyperostosis patterns between CED1 and CED2, suggest distinct functions between TGFB1 and TGFB2 in human skeletal development and homeostasis.
TGFB2 的潜伏相关肽域的紧身衣区的杂合突变导致卡姆拉蒂-恩格尔曼病 II 型。
卡穆拉蒂-恩格尔曼病(Camurati-Engelmann disease,CED)是一种常染色体显性骨发育不良症,其特征是颅底和长骨骺端进行性骨质增生。根据是否存在 TGFB1 突变,CED 又分为两个亚型,即 CED1 和 CED2。在本研究中,我们利用外显子组测序技术调查了三个血统中 CED2 的遗传原因,并在三名患者中发现了两个 TGFB2 基因的新发杂合突变。这两个突变都位于编码原 TGF-β2 的潜伏相关肽(LAP)的紧身衣亚域的基因区域。对突变 LAP 的结构模拟表明,突变可能会导致构象发生重大变化,并导致 TGF-β2 失活能力下降。活性测定证实,TGF-β2/SMAD 信号传导明显增加。使用其中一名CED2患者的iPS细胞进行的体外成骨分化实验显示,骨化明显增强,这表明CED2的致病机制是LAP功能缺失导致TGF-β2激活增加。这些结果与 CED1 和 CED2 骨质疏松模式的差异相结合,表明 TGFB1 和 TGFB2 在人类骨骼发育和稳态中具有不同的功能。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


