Ab Initio Potentials for the Ground S0 and the First Electronically Excited Singlet S1 States of Benzene-Helium with Application to Tunneling Intermolecular Vibrational States.
{"title":"<i>Ab Initio</i> Potentials for the Ground <i>S</i><sub>0</sub> and the First Electronically Excited Singlet <i>S</i><sub>1</sub> States of Benzene-Helium with Application to Tunneling Intermolecular Vibrational States.","authors":"Leonid Shirkov","doi":"10.1021/acs.jpca.4c01491","DOIUrl":null,"url":null,"abstract":"<p><p>We present new <i>ab initio</i> intermolecular potential energy surfaces for the benzene-helium complex in its ground (<i>S</i><sub>0</sub>) and first excited (<i>S</i><sub>1</sub>) states. The coupled-cluster level of theory with single, double, and perturbative triple excitations, CCSD(T), was used to calculate the ground state potential. The excited state potential was obtained by adding the excitation energies <i>S</i><sub>0</sub> → <i>S</i><sub>1</sub> of the complex, calculated using the equation of motion approach EOM-CCSD, to the ground state potential interaction energies. Analytical potentials are constructed and applied to study the structural and vibrational dynamics of benzene-helium. The binding energies and equilibrium distances of the ground and excited states were found to be 89 cm<sup>-1</sup>, 3.14 Å and 77 cm<sup>-1</sup>, 3.20 Å, respectively. The calculated vibrational energy levels exhibit tunneling of He through the benzene plane and are in reasonable agreement with recently reported experimental values for both the ground and excited states [Hayashi, M.; Ohshima, Y. <i>J. Phys. Chem. Lett.</i> <b>2020</b>, <i>11</i>, 9745]. Prospects for the theoretical study of complexes with large aromatic molecules and He are also discussed.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":null,"pages":null},"PeriodicalIF":2.7000,"publicationDate":"2024-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11299187/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpca.4c01491","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/17 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
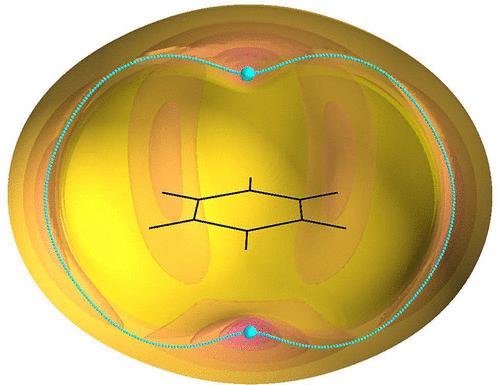
We present new ab initio intermolecular potential energy surfaces for the benzene-helium complex in its ground (S0) and first excited (S1) states. The coupled-cluster level of theory with single, double, and perturbative triple excitations, CCSD(T), was used to calculate the ground state potential. The excited state potential was obtained by adding the excitation energies S0 → S1 of the complex, calculated using the equation of motion approach EOM-CCSD, to the ground state potential interaction energies. Analytical potentials are constructed and applied to study the structural and vibrational dynamics of benzene-helium. The binding energies and equilibrium distances of the ground and excited states were found to be 89 cm-1, 3.14 Å and 77 cm-1, 3.20 Å, respectively. The calculated vibrational energy levels exhibit tunneling of He through the benzene plane and are in reasonable agreement with recently reported experimental values for both the ground and excited states [Hayashi, M.; Ohshima, Y. J. Phys. Chem. Lett.2020, 11, 9745]. Prospects for the theoretical study of complexes with large aromatic molecules and He are also discussed.
苯氦的地面 S0 态和首次电子激发单态 S1 态的 Ab Initio 电位及其在分子间振动隧穿状态中的应用。
我们提出了苯-氦复合物在基态(S0)和第一激发态(S1)时的新的非初始分子间势能面。计算基态势能时使用了单激发、双激发和扰动三激发的耦合簇理论水平 CCSD(T)。激发态电势是通过将使用 EOM-CCSD 运动方程方法计算的复合物激发能量 S0 → S1 与基态电势相互作用能量相加而得到的。构建了分析势,并将其用于研究苯-氦的结构和振动动力学。结果发现,基态和激发态的结合能和平衡距离分别为 89 cm-1, 3.14 Å 和 77 cm-1, 3.20 Å。计算出的振动能级显示了 He 通过苯平面的隧道效应,与最近报告的基态和激发态的实验值基本一致[Hayashi, M.; Ohshima, Y. J. Phys. Chem. Lett.]此外,还讨论了理论研究芳香族大分子与 He 的配合物的前景。
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


