{"title":"Genotyping and Molecular Characterization of VP6 and NSP4 Genes of Unusual Rotavirus Group A Isolated from Children with Acute Gastroenteritis.","authors":"Charilaos Dellis, Elizabeth-Barbara Tatsi, Dimitra-Maria Koukou, Filippos Filippatos, Evangelia-Eirini Vetouli, Emmanouil Zoumakis, Athanasios Michos, Vasiliki Syriopoulou","doi":"10.1155/2024/3263228","DOIUrl":null,"url":null,"abstract":"<p><p>Group A rotavirus (RVA), which causes acute gastroenteritis (AGE) in children worldwide, is categorized mainly based on VP7 (genotype G) and VP4 (genotype P) genes. Genotypes that circulate at <1% are considered unusual. Important genes also include VP6 (genotype I) and NSP4 (genotype E). VP6 establishes the group and affects immunogenicity, while NSP4, as an enterotoxin, is responsible for the clinical symptoms. The aim of this study was to genotype the VP6 and NSP4 genes and molecularly characterize the NSP4 and VP6 genes of unusual RVA. Unusual RVA strains extracted from fecal samples of children ≤16 years with AGE were genotyped in VP6 and NSP4 genes with Sanger sequencing. In a 15-year period (2007-2021), 54.8% (34/62) of unusual RVA were successfully I and E genotyped. Three different I and E genotypes were identified; I2 (73.5%, 25/34) and E2 (35.3%, 12/34) were the most common. E3 genotype was detected from 2017 onwards. The uncommon combination of I2-E3 was found in 26.5% (9/34) of the strains and G3-P[9]-I2-E3 remained the most frequent G-P-I-E combination (20.6%, 7/34). Children infected with RVA E2 strains had a statistically higher frequency of dehydration (50%) than those infected with RVA E3 strains (<i>p</i> = 0.019). Multiple substitutions were detected in NSP4, but their functional effect remains unknown. The result indicates the genetic diversity of RVA strains. Continuous surveillance of the RVA based on the whole genome will provide better knowledge of its evolution.</p>","PeriodicalId":7473,"journal":{"name":"Advances in Virology","volume":"2024 ","pages":"3263228"},"PeriodicalIF":1.4000,"publicationDate":"2024-07-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11239230/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advances in Virology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/3263228","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"VIROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
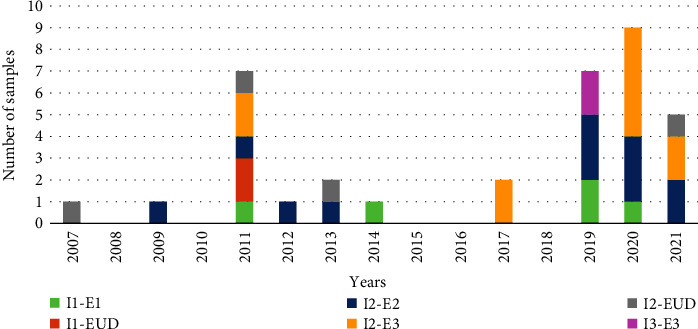
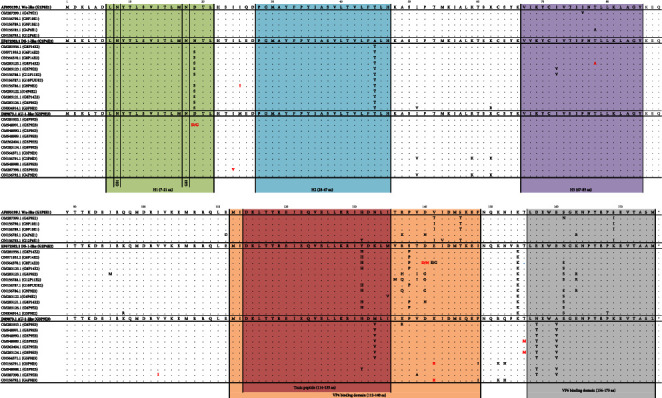
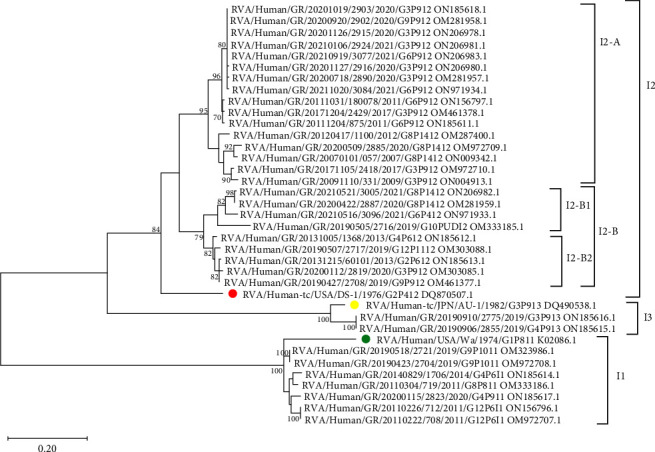
Group A rotavirus (RVA), which causes acute gastroenteritis (AGE) in children worldwide, is categorized mainly based on VP7 (genotype G) and VP4 (genotype P) genes. Genotypes that circulate at <1% are considered unusual. Important genes also include VP6 (genotype I) and NSP4 (genotype E). VP6 establishes the group and affects immunogenicity, while NSP4, as an enterotoxin, is responsible for the clinical symptoms. The aim of this study was to genotype the VP6 and NSP4 genes and molecularly characterize the NSP4 and VP6 genes of unusual RVA. Unusual RVA strains extracted from fecal samples of children ≤16 years with AGE were genotyped in VP6 and NSP4 genes with Sanger sequencing. In a 15-year period (2007-2021), 54.8% (34/62) of unusual RVA were successfully I and E genotyped. Three different I and E genotypes were identified; I2 (73.5%, 25/34) and E2 (35.3%, 12/34) were the most common. E3 genotype was detected from 2017 onwards. The uncommon combination of I2-E3 was found in 26.5% (9/34) of the strains and G3-P[9]-I2-E3 remained the most frequent G-P-I-E combination (20.6%, 7/34). Children infected with RVA E2 strains had a statistically higher frequency of dehydration (50%) than those infected with RVA E3 strains (p = 0.019). Multiple substitutions were detected in NSP4, but their functional effect remains unknown. The result indicates the genetic diversity of RVA strains. Continuous surveillance of the RVA based on the whole genome will provide better knowledge of its evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


