Enantioselective alkyl–alkyl coupling by Ni-catalysed asymmetric cross-hydrodimerization of alkenes
0 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
Saturated tertiary stereogenic carbon centres are common in small molecules and organic materials. Transition-metal-catalysed asymmetric alkyl–alkyl bond formation processes represent contemporary techniques for the straightforward and efficient construction of saturated tertiary stereogenic carbon centres. However, reaction modes for asymmetric alkyl–alkyl bond formation between sp3-hybridized carbon atoms, C(sp3)–C(sp3), are limited yet highly desirable. Here a mode for asymmetric alkyl–alkyl bond formation enabled by Ni-catalysed asymmetric alkyl–alkyl cross-coupling between alkenes has been developed to construct tertiary stereogenic carbon centres. Ni-catalysed asymmetric cross-hydrodimerization of N-acyl enamines and unactivated alkenes enables head-to-tail regioselectivity and excellent levels of chemo- and enantioselectivity. Notably, the reaction proceeds in the presence of both reducing and oxidizing reagents, rendering alkenes as the sole precursors to forge enantioselective alkyl–alkyl bonds. The exclusive head-to-tail cross-hydrodimerization of distinct alkenes opens the way to access saturated tertiary stereogenic carbon centres from alkenes. Methods for asymmetric alkyl–alkyl bond formation between sp3-hybridized carbon atoms, C(sp3)–C(sp3), are limited yet highly desirable. Now an approach for asymmetric alkyl–alkyl bond formation by Ni-catalysed cross-coupling between alkenes has been developed to construct tertiary stereogenic carbon centres with head-to-tail regioselectivity and excellent chemo- and enantioselectivity.
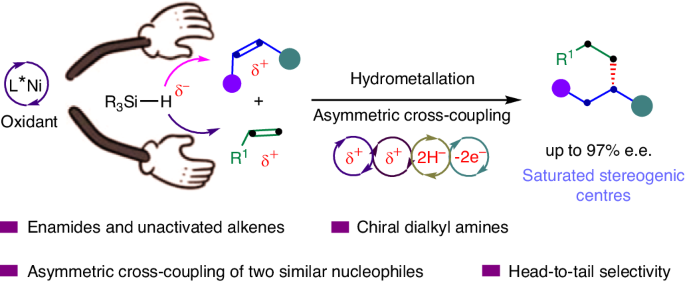
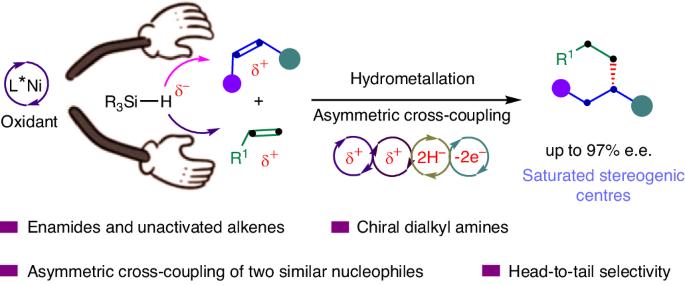
通过镍催化的烯烃不对称交叉二聚体化实现对映选择性烷基-烷基偶联
饱和三级立体碳中心在小分子和有机材料中很常见。过渡金属催化的不对称烷基-烷基键形成过程是直接高效构建饱和三级立体碳中心的现代技术。然而,在 sp3 杂化碳原子(C(sp3)-C(sp3))之间形成不对称烷基-烷基键的反应模式非常有限,但却非常理想。在此,我们开发了一种通过镍催化的烯烃间不对称烷基-烷基交叉偶联形成不对称烷基-烷基键的模式,以构建三级立体碳中心。镍催化的 N-酰基烯胺和未活化烯烃的不对称交叉二聚化反应具有从头到尾的区域选择性以及出色的化学和对映选择性。值得注意的是,该反应在存在还原和氧化试剂的情况下进行,使烯成为形成对映选择性烷基-烷基键的唯一前体。独特的烯烃头尾交叉氢二聚化为从烯烃中获得饱和的三级立体碳中心开辟了道路。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


