{"title":"Spatiotemporal and Species-Crossing Transmission Dynamics of Subclade 2.3.4.4b H5Nx HPAIVs","authors":"Minghui Li, Jingman Tian, Xiaoli Bai, Xingdong Song, Zhiguo Zhao, Jianzhong Shi, Guohua Deng, Xianying Zeng, Guobin Tian, Huihui Kong, Jinxiong Liu, Chengjun Li, Yanbing Li","doi":"10.1155/2024/2862053","DOIUrl":null,"url":null,"abstract":"<div>\n <p>Subclade 2.3.4.4b H5Nx highly pathogenic avian influenza (HPAI) viruses, emerged in 2013 with multiple subtypes of H5N8, H5N1, and H5N6, had unprecedently caused a global epizootic by H5N1 since 2021, which had devasted multiple species of wild birds, poultry, and wild mammals (terrestrial and marine) with a high mortality, causing severe ecological damage. The infected wild mammals may become new “mixers” for influenza viruses, posing the potential transmission to human. Frequent outbreaks of subclade 2.3.4.4b H5Nx viruses among wild birds and poultry had exposed major gaps in our knowledge on their evolution, spatiotemporal diffusion, and species-crossing transmission. Here, we integrated the phylogenetic and epidemiological data of subclade 2.3.4.4b H5Nx viruses in public database and used Bayesian phylodynamic analysis to reveal the pattern of the global large-scale transmission. Phylogenic analysis demonstrated that the HA gene of these viruses diverged into two dominant clusters around 2015 and 2016. The Bayesian phylodynamic analysis illustrated that the viruses presented spatiotemporally complex transmission network with geographical and host relative expansion and recombination with different subtypes of NA segment. Spatially, the Russian Federation (Siberia) was identified as the primary hub for virus transmission, which was further facilitated by the establishment of strong epidemiological linkages between West Europe and broader regions, such as North America. As for hosts, wild Anseriformes were the primary species for the virus spillover, contributing to the spatial expansion and rapid diffusion globally of subclade 2.3.4.4b viruses. We investigated the phylogeny of subclade 2.3.4.4b H5Nx viruses and the spatiotemporal pattern of transmission with initial location and the primary host, which could provide comprehensive insights for subclade 2.3.4.4b H5Nx viruses. Due to the wild birds involved the widespread of subclade 2.3.4.4b H5Nx viruses, the epizootics in poultry are inevitable, so we highly recommend to apply the policy of culling plus with vaccination to protect the poultry industry and potentially protect the public health.</p>\n </div>","PeriodicalId":234,"journal":{"name":"Transboundary and Emerging Diseases","volume":"2024 1","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2024-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/2024/2862053","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Transboundary and Emerging Diseases","FirstCategoryId":"97","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/2024/2862053","RegionNum":2,"RegionCategory":"农林科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"INFECTIOUS DISEASES","Score":null,"Total":0}
引用次数: 0
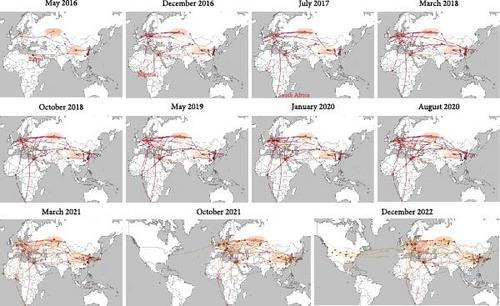
Abstract
Subclade 2.3.4.4b H5Nx highly pathogenic avian influenza (HPAI) viruses, emerged in 2013 with multiple subtypes of H5N8, H5N1, and H5N6, had unprecedently caused a global epizootic by H5N1 since 2021, which had devasted multiple species of wild birds, poultry, and wild mammals (terrestrial and marine) with a high mortality, causing severe ecological damage. The infected wild mammals may become new “mixers” for influenza viruses, posing the potential transmission to human. Frequent outbreaks of subclade 2.3.4.4b H5Nx viruses among wild birds and poultry had exposed major gaps in our knowledge on their evolution, spatiotemporal diffusion, and species-crossing transmission. Here, we integrated the phylogenetic and epidemiological data of subclade 2.3.4.4b H5Nx viruses in public database and used Bayesian phylodynamic analysis to reveal the pattern of the global large-scale transmission. Phylogenic analysis demonstrated that the HA gene of these viruses diverged into two dominant clusters around 2015 and 2016. The Bayesian phylodynamic analysis illustrated that the viruses presented spatiotemporally complex transmission network with geographical and host relative expansion and recombination with different subtypes of NA segment. Spatially, the Russian Federation (Siberia) was identified as the primary hub for virus transmission, which was further facilitated by the establishment of strong epidemiological linkages between West Europe and broader regions, such as North America. As for hosts, wild Anseriformes were the primary species for the virus spillover, contributing to the spatial expansion and rapid diffusion globally of subclade 2.3.4.4b viruses. We investigated the phylogeny of subclade 2.3.4.4b H5Nx viruses and the spatiotemporal pattern of transmission with initial location and the primary host, which could provide comprehensive insights for subclade 2.3.4.4b H5Nx viruses. Due to the wild birds involved the widespread of subclade 2.3.4.4b H5Nx viruses, the epizootics in poultry are inevitable, so we highly recommend to apply the policy of culling plus with vaccination to protect the poultry industry and potentially protect the public health.
期刊介绍:
Transboundary and Emerging Diseases brings together in one place the latest research on infectious diseases considered to hold the greatest economic threat to animals and humans worldwide. The journal provides a venue for global research on their diagnosis, prevention and management, and for papers on public health, pathogenesis, epidemiology, statistical modeling, diagnostics, biosecurity issues, genomics, vaccine development and rapid communication of new outbreaks. Papers should include timely research approaches using state-of-the-art technologies. The editors encourage papers adopting a science-based approach on socio-economic and environmental factors influencing the management of the bio-security threat posed by these diseases, including risk analysis and disease spread modeling. Preference will be given to communications focusing on novel science-based approaches to controlling transboundary and emerging diseases. The following topics are generally considered out-of-scope, but decisions are made on a case-by-case basis (for example, studies on cryptic wildlife populations, and those on potential species extinctions):
Pathogen discovery: a common pathogen newly recognised in a specific country, or a new pathogen or genetic sequence for which there is little context about — or insights regarding — its emergence or spread.
Prevalence estimation surveys and risk factor studies based on survey (rather than longitudinal) methodology, except when such studies are unique. Surveys of knowledge, attitudes and practices are within scope.
Diagnostic test development if not accompanied by robust sensitivity and specificity estimation from field studies.
Studies focused only on laboratory methods in which relevance to disease emergence and spread is not obvious or can not be inferred (“pure research” type studies).
Narrative literature reviews which do not generate new knowledge. Systematic and scoping reviews, and meta-analyses are within scope.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


