Michael K Gilson, Lawrence E Stewart, Michael J Potter, Simon P Webb
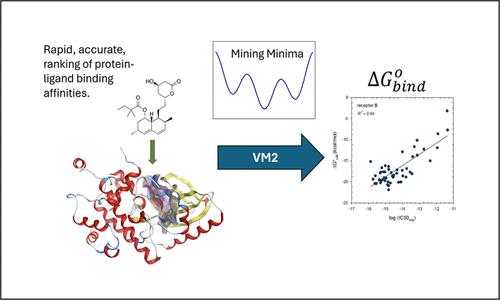
{"title":"Rapid, Accurate, Ranking of Protein-Ligand Binding Affinities with VM2, the Second-Generation Mining Minima Method.","authors":"Michael K Gilson, Lawrence E Stewart, Michael J Potter, Simon P Webb","doi":"10.1021/acs.jctc.4c00407","DOIUrl":null,"url":null,"abstract":"<p><p>The structure-based technologies most widely used to rank the affinities of candidate small molecule drugs for proteins range from faster but less reliable docking methods to slower but more accurate explicit solvent free energy methods. In recent years, we have advanced another technology, which is called mining minima because it \"mines\" out the main contributions to the chemical potentials of the free and bound molecular species by identifying and characterizing their main local energy minima. The present study provides systematic benchmarks of the accuracy and computational speed of mining minima, as implemented in the VeraChem Mining Minima Generation 2 (VM2) code, across two well-regarded protein-ligand benchmark data sets, for which there are already benchmark data for docking, free energy, and other computational methods. A core result is that VM2's accuracy approaches that of explicit solvent free energy methods at a far lower computational cost. In finer-grained analyses, we also examine the influence of various run settings, such as the treatment of crystallographic water molecules, on the accuracy, and define the costs in time and dollars of representative runs on Amazon Web Services (AWS) compute instances with various CPU and GPU combinations. We also use the benchmark data to determine the importance of VM2's correction from generalized Born to finite-difference Poisson-Boltzmann results for each energy well and find that this correction affords a remarkably consistent improvement in accuracy at a modest computational cost. The present results establish VM2 as a distinctive technology for early-stage drug discovery, which provides a strong combination of efficiency and predictivity.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00407","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/11 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
The structure-based technologies most widely used to rank the affinities of candidate small molecule drugs for proteins range from faster but less reliable docking methods to slower but more accurate explicit solvent free energy methods. In recent years, we have advanced another technology, which is called mining minima because it "mines" out the main contributions to the chemical potentials of the free and bound molecular species by identifying and characterizing their main local energy minima. The present study provides systematic benchmarks of the accuracy and computational speed of mining minima, as implemented in the VeraChem Mining Minima Generation 2 (VM2) code, across two well-regarded protein-ligand benchmark data sets, for which there are already benchmark data for docking, free energy, and other computational methods. A core result is that VM2's accuracy approaches that of explicit solvent free energy methods at a far lower computational cost. In finer-grained analyses, we also examine the influence of various run settings, such as the treatment of crystallographic water molecules, on the accuracy, and define the costs in time and dollars of representative runs on Amazon Web Services (AWS) compute instances with various CPU and GPU combinations. We also use the benchmark data to determine the importance of VM2's correction from generalized Born to finite-difference Poisson-Boltzmann results for each energy well and find that this correction affords a remarkably consistent improvement in accuracy at a modest computational cost. The present results establish VM2 as a distinctive technology for early-stage drug discovery, which provides a strong combination of efficiency and predictivity.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


