Fariya Khan, Altaf Ahmad Shah, Ajay Kumar, Salman Akhtar
{"title":"In Silico Investigation against Inhibitors of Alpha-Amylase Using Structure-based Screening, Molecular Docking, and Molecular Simulations Studies.","authors":"Fariya Khan, Altaf Ahmad Shah, Ajay Kumar, Salman Akhtar","doi":"10.1007/s12013-024-01403-9","DOIUrl":null,"url":null,"abstract":"<p><p>Type-II diabetes mellitus is a chronic disorder that results from fluctuations in the glucose level leading to hyperglycemia with severe adverse effects increasing worldwide. Alpha-Amylase is the key enzyme involved in the mechanism of glucose formation therefore Alpha-Amylase inhibitors have become a therapeutic target in the development of new leads as they have the potential to suppress glucose levels. Existing drugs targeting Alpha-Amylase highlight major drawbacks in terms of poor absorption rate that causes several gastrointestinal issues. So, this research is aimed to develop novel inhibitors interacting with Alpha-Amylase's active site using structural-based screening, binding pattern analysis, and molecular dynamic simulation. Hence, to search for a potential lead, we analyzed a total of 133 valiolamine derivatives and 535 desoxynojirimycin derivatives that exhibited drug-like properties screened through Lipinski filters. Virtual screening followed by binding interaction analysis we identified ten compounds that exhibited better binding energy scores compared to the standard drugs voglibose and miglitol, used in our study. The docking analysis, ADMET and metabolic site prediction estimated the best top two compounds with good drug profiles. Further, top compounds VG9 and VG15 were promoted to simulation study using the Biovia Discovery study to access the stability at a time interval of 100 ns. MD simulation results revealed that our compound VG9 possesses better conformational stability in the complex to the active site residues of Alpha-Amylase target protein than standard drug voglibose. Thus, our investigation revealed that compound VG9 also exhibits the best pharmacokinetic as well as binding affinity results and could act as a potential lead compound targeting Alpha-Amylase for Type II diabetes.</p>","PeriodicalId":510,"journal":{"name":"Cell Biochemistry and Biophysics","volume":null,"pages":null},"PeriodicalIF":1.8000,"publicationDate":"2024-07-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Biochemistry and Biophysics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12013-024-01403-9","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
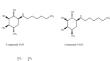
Type-II diabetes mellitus is a chronic disorder that results from fluctuations in the glucose level leading to hyperglycemia with severe adverse effects increasing worldwide. Alpha-Amylase is the key enzyme involved in the mechanism of glucose formation therefore Alpha-Amylase inhibitors have become a therapeutic target in the development of new leads as they have the potential to suppress glucose levels. Existing drugs targeting Alpha-Amylase highlight major drawbacks in terms of poor absorption rate that causes several gastrointestinal issues. So, this research is aimed to develop novel inhibitors interacting with Alpha-Amylase's active site using structural-based screening, binding pattern analysis, and molecular dynamic simulation. Hence, to search for a potential lead, we analyzed a total of 133 valiolamine derivatives and 535 desoxynojirimycin derivatives that exhibited drug-like properties screened through Lipinski filters. Virtual screening followed by binding interaction analysis we identified ten compounds that exhibited better binding energy scores compared to the standard drugs voglibose and miglitol, used in our study. The docking analysis, ADMET and metabolic site prediction estimated the best top two compounds with good drug profiles. Further, top compounds VG9 and VG15 were promoted to simulation study using the Biovia Discovery study to access the stability at a time interval of 100 ns. MD simulation results revealed that our compound VG9 possesses better conformational stability in the complex to the active site residues of Alpha-Amylase target protein than standard drug voglibose. Thus, our investigation revealed that compound VG9 also exhibits the best pharmacokinetic as well as binding affinity results and could act as a potential lead compound targeting Alpha-Amylase for Type II diabetes.
期刊介绍:
Cell Biochemistry and Biophysics (CBB) aims to publish papers on the nature of the biochemical and biophysical mechanisms underlying the structure, control and function of cellular systems
The reports should be within the framework of modern biochemistry and chemistry, biophysics and cell physiology, physics and engineering, molecular and structural biology. The relationship between molecular structure and function under investigation is emphasized.
Examples of subject areas that CBB publishes are:
· biochemical and biophysical aspects of cell structure and function;
· interactions of cells and their molecular/macromolecular constituents;
· innovative developments in genetic and biomolecular engineering;
· computer-based analysis of tissues, cells, cell networks, organelles, and molecular/macromolecular assemblies;
· photometric, spectroscopic, microscopic, mechanical, and electrical methodologies/techniques in analytical cytology, cytometry and innovative instrument design
For articles that focus on computational aspects, authors should be clear about which docking and molecular dynamics algorithms or software packages are being used as well as details on the system parameterization, simulations conditions etc. In addition, docking calculations (virtual screening, QSAR, etc.) should be validated either by experimental studies or one or more reliable theoretical cross-validation methods.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


