{"title":"The mechanical properties of advanced U–Si compounds using first principles method","authors":"Hengfeng Gong, Daxi Guo, Jianhan Zhai, Jiwei Wang, Minzhou Chen, Lixiang Wu, Jiaxiang Xue, Yehong Liao","doi":"10.1140/epjb/s10051-024-00736-x","DOIUrl":null,"url":null,"abstract":"<div><p>Due to high thermal conductivity and higher fissile density, one of the most options being pursued for accident-tolerant fuels is U–Si fuels. Unfortunately, the data available for U–Si fuels are rather limited now. Based on a few assumptions regarding geometry structures, electronic properties, elastic constants and hardness are predicted systematically for U–Si compounds using density functional theory (DFT) in this work. The calculation results show that the U<sub>3</sub>Si<sub>2</sub>, β-U<sub>3</sub>Si and γ-U<sub>3</sub>Si compounds are metallic and brittle, which is in good agreement with the previous experimental results. The Si-rich USi<sub>1.84</sub> compound presents the ductile. By analyzing shear anisotropy factors, it is found that the U<sub>3</sub>Si<sub>2</sub>, β-U<sub>3</sub>Si and Si-rich USi<sub>1.84</sub> have the anisotropic characteristics, while the γ-U<sub>3</sub>Si is isotropic. Moreover, among the U<sub>3</sub>Si<sub>2</sub>, β-U<sub>3</sub>Si, γ-U<sub>3</sub>Si and Si-rich USi<sub>1.84</sub> compounds, the γ-U<sub>3</sub>Si is hardest and Si-rich USi<sub>1.84</sub> is softest, then U<sub>3</sub>Si<sub>2</sub> is much harder than that of β-U<sub>3</sub>Si by the Voigt hardness calculations. The hardness value of U<sub>3</sub>Si<sub>2</sub> is overestimated in our calculation than experimental data, mainly because the model may be ideal without considering defects and microstructure. The theoretical investigation of this work will be expected to provide parameters for the physical models in the advanced fuel rod performance analysis.</p><h3>Graphical abstract</h3>\n<div><figure><div><div><picture><source><img></source></picture></div><div><p>The relationship between crystal structures and three dimensions elastic constants</p></div></div></figure></div></div>","PeriodicalId":787,"journal":{"name":"The European Physical Journal B","volume":"97 7","pages":""},"PeriodicalIF":1.6000,"publicationDate":"2024-07-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The European Physical Journal B","FirstCategoryId":"4","ListUrlMain":"https://link.springer.com/article/10.1140/epjb/s10051-024-00736-x","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
引用次数: 0
Abstract
Due to high thermal conductivity and higher fissile density, one of the most options being pursued for accident-tolerant fuels is U–Si fuels. Unfortunately, the data available for U–Si fuels are rather limited now. Based on a few assumptions regarding geometry structures, electronic properties, elastic constants and hardness are predicted systematically for U–Si compounds using density functional theory (DFT) in this work. The calculation results show that the U3Si2, β-U3Si and γ-U3Si compounds are metallic and brittle, which is in good agreement with the previous experimental results. The Si-rich USi1.84 compound presents the ductile. By analyzing shear anisotropy factors, it is found that the U3Si2, β-U3Si and Si-rich USi1.84 have the anisotropic characteristics, while the γ-U3Si is isotropic. Moreover, among the U3Si2, β-U3Si, γ-U3Si and Si-rich USi1.84 compounds, the γ-U3Si is hardest and Si-rich USi1.84 is softest, then U3Si2 is much harder than that of β-U3Si by the Voigt hardness calculations. The hardness value of U3Si2 is overestimated in our calculation than experimental data, mainly because the model may be ideal without considering defects and microstructure. The theoretical investigation of this work will be expected to provide parameters for the physical models in the advanced fuel rod performance analysis.
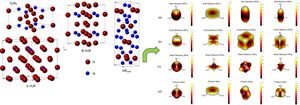
Graphical abstract
The relationship between crystal structures and three dimensions elastic constants

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


