Chemoenzymatic total synthesis of alchivemycin A
0 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
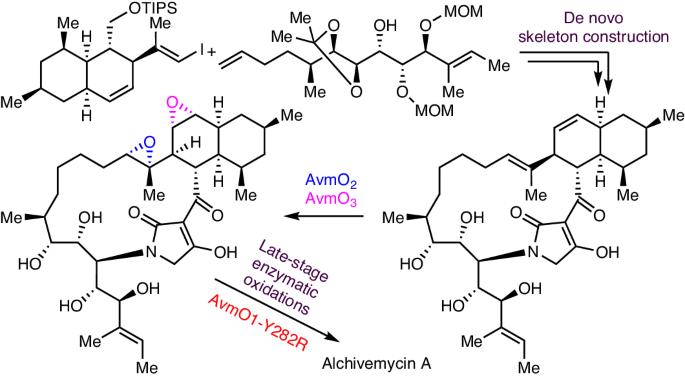
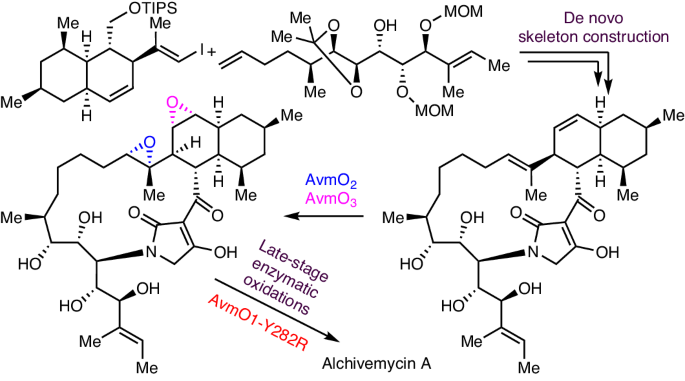
Alchivemycin A belongs to a unique class of polyketide natural products isolated from plant-derived actinomycete Streptomyces. It shows potent antibacterial activity and anti-tumour activity. However, its inherent structural complexity and high oxidation state, especially the 2H-tetrahydro-4,6-dioxo-1,2-oxazine (TDO) ring system, present synthetic challenges. Here we report the total synthesis of alchivemycin A using a chemoenzymatic approach that combines de novo skeleton construction and late-stage enzymatic oxidation reactions. The convergent synthesis of the highly functionalized unnatural tetramic acid-bearing intermediate is achieved by boron-alkyl Suzuki−Miyaura cross-coupling, macrolactamization and Lacey–Dieckmann condensation reactions. Efficient enzymatic epoxidations using the redox enzymes AvmO3 and AvmO2 allow rapid access to the desired diepoxide product regio- and stereoselectively. Subsequently, a flavin adenine dinucleotide-dependent enzyme AvmO1 variant optimized via rational protein engineering, AvmO1-Y282R, was used to convert the tetramic acid ring into the TDO ring through a Baeyer–Villiger-type transformation, completing the chemoenzymatic synthesis of alchivemycin A. This work paves the way to further explore the biological functions of alchivemycin A and highlights the utility of chemoenzymatic strategies to tackle synthetic challenges in complex molecule synthesis. The asymmetric total synthesis of alchivemycin A is reported. The synthesis uses a chemoenzymatic approach that comprises de novo skeleton construction and a late-stage enzymatic oxidation cascade.
化学酶法全合成茜草霉素 A
Alchivemycin A 属于一类独特的多酮类天然产品,从植物源放线菌链霉菌中分离出来。它具有很强的抗菌活性和抗肿瘤活性。然而,其固有的结构复杂性和高氧化态,尤其是 2H-四氢-4,6-二氧代-1,2-恶嗪(TDO)环系统,给合成带来了挑战。在此,我们报告了利用化学酶法结合新骨架构建和后期酶促氧化反应,全合成茜草霉素 A 的过程。通过硼烷基铃木-米亚乌拉交叉偶联反应、大内酰胺化反应和莱西-迪克曼缩合反应,实现了高度官能化的非天然四酸中间体的聚合合成。利用氧化还原酶 AvmO3 和 AvmO2 进行高效的酶环氧化反应,可以迅速获得所需的二环氧化物产品的区域和立体选择性。随后,通过合理蛋白质工程优化的黄素腺嘌呤二核苷酸依赖酶AvmO1变体AvmO1-Y282R通过拜耳-维利格型转化将四元酸环转化为TDO环,完成了阿奇霉素A的化学酶法合成。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


