Loss of Kmt2c or Kmt2d drives brain metastasis via KDM6A-dependent upregulation of MMP3
IF 19.1
1区 生物学
Q1 CELL BIOLOGY
引用次数: 0
Abstract
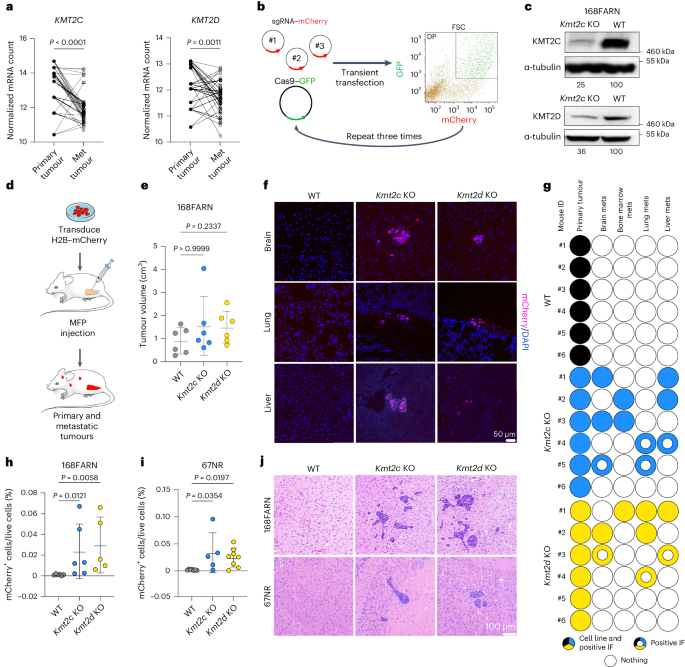
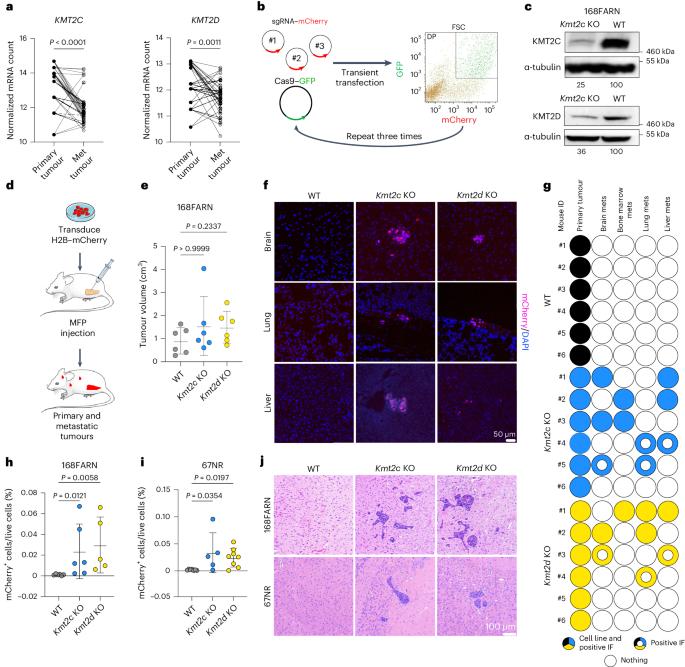
KMT2C and KMT2D, encoding histone H3 lysine 4 methyltransferases, are among the most commonly mutated genes in triple-negative breast cancer (TNBC). However, how these mutations may shape epigenomic and transcriptomic landscapes to promote tumorigenesis is largely unknown. Here we describe that deletion of Kmt2c or Kmt2d in non-metastatic murine models of TNBC drives metastasis, especially to the brain. Global chromatin profiling and chromatin immunoprecipitation followed by sequencing revealed altered H3K4me1, H3K27ac and H3K27me3 chromatin marks in knockout cells and demonstrated enhanced binding of the H3K27me3 lysine demethylase KDM6A, which significantly correlated with gene expression. We identified Mmp3 as being commonly upregulated via epigenetic mechanisms in both knockout models. Consistent with these findings, samples from patients with KMT2C-mutant TNBC have higher MMP3 levels. Downregulation or pharmacological inhibition of KDM6A diminished Mmp3 upregulation induced by the loss of histone–lysine N-methyltransferase 2 (KMT2) and prevented brain metastasis similar to direct downregulation of Mmp3. Taken together, we identified the KDM6A–matrix metalloproteinase 3 axis as a key mediator of KMT2C/D loss-driven metastasis in TNBC. Seehawer et al. show that deletion of Kmt2c or Kmt2d promotes brain metastasis in mouse models of triple-negative breast cancer due to altered KDM6A activity and upregulated MMP3 expression, which may constitute a potential therapeutic target.
Kmt2c 或 Kmt2d 的缺失通过 KDM6A 依赖性上调 MMP3 推动脑转移
编码组蛋白 H3 赖氨酸 4 甲基转移酶的 KMT2C 和 KMT2D 是三阴性乳腺癌(TNBC)中最常见的突变基因之一。然而,这些突变如何形成表观基因组和转录组景观以促进肿瘤发生,目前还不清楚。在这里,我们描述了在非转移性小鼠 TNBC 模型中缺失 Kmt2c 或 Kmt2d 会导致肿瘤转移,尤其是向脑部转移。全染色质图谱分析和染色质免疫共沉淀测序显示,基因敲除细胞中的H3K4me1、H3K27ac和H3K27me3染色质标记发生了改变,并证明H3K27me3赖氨酸去甲基化酶KDM6A的结合增强,这与基因表达显著相关。我们发现在这两种基因敲除模型中,Mmp3通常通过表观遗传机制上调。与这些发现一致的是,KMT2C突变TNBC患者样本中的MMP3水平较高。KDM6A的下调或药理抑制降低了组蛋白-赖氨酸N-甲基转移酶2(KMT2)缺失诱导的Mmp3上调,并防止了脑转移,这与直接下调Mmp3类似。综上所述,我们发现KDM6A-基质金属蛋白酶3轴是KMT2C/D缺失驱动TNBC转移的关键介质。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Cell Biology
生物-细胞生物学
CiteScore
28.40
自引率
0.90%
发文量
219
审稿时长
3 months
期刊介绍:
Nature Cell Biology, a prestigious journal, upholds a commitment to publishing papers of the highest quality across all areas of cell biology, with a particular focus on elucidating mechanisms underlying fundamental cell biological processes. The journal's broad scope encompasses various areas of interest, including but not limited to:
-Autophagy
-Cancer biology
-Cell adhesion and migration
-Cell cycle and growth
-Cell death
-Chromatin and epigenetics
-Cytoskeletal dynamics
-Developmental biology
-DNA replication and repair
-Mechanisms of human disease
-Mechanobiology
-Membrane traffic and dynamics
-Metabolism
-Nuclear organization and dynamics
-Organelle biology
-Proteolysis and quality control
-RNA biology
-Signal transduction
-Stem cell biology
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


