Catalyzing precision: unraveling the diagnostic conundrum of tunisian familial hypophosphatasia case through integrative clinical and molecular approaches.
Yessine Amri, Rym Dabboubi, Monia Khemiri, Elham Jebabli, Sondess Hadj Fredj, Sarra Ben Ahmed, Yosr Jouini, Faida Ouali, Taieb Messaoud
{"title":"Catalyzing precision: unraveling the diagnostic conundrum of tunisian familial hypophosphatasia case through integrative clinical and molecular approaches.","authors":"Yessine Amri, Rym Dabboubi, Monia Khemiri, Elham Jebabli, Sondess Hadj Fredj, Sarra Ben Ahmed, Yosr Jouini, Faida Ouali, Taieb Messaoud","doi":"10.1007/s00438-024-02157-y","DOIUrl":null,"url":null,"abstract":"<p><p>Familial Hypophosphatasia presents a complex diagnostic challenge due to its wide-ranging clinical manifestations and genetic heterogeneity. This study aims to elucidate the molecular underpinnings of familial Hypophosphatasia within a Tunisian family harboring a rare c.896 T > C mutation in the ALPL gene, offering insights into genotype-phenotype correlations and potential therapeutic avenues. The study employs a comprehensive approach, integrating biochemical examination, genetic analysis, structural modeling, and functional insights to unravel the impact of this rare mutation. Genetic investigation revealed the presence of the p.Leu299Pro mutation within the ALPL gene in affected family members. This mutation is strategically positioned in proximity to both the catalytic site and the metal-binding domain, suggesting potential functional consequences. Homology modeling techniques were employed to predict the 3D structure of TNSALP, providing insights into the structural context of the mutation. Our findings suggest that the mutation may induce conformational changes in the vicinity of the catalytic site and metal-binding domain, potentially affecting substrate recognition and catalytic efficiency. Molecular dynamics simulations were instrumental in elucidating the dynamic behavior of the tissue-nonspecific alkaline phosphatase isozyme (TNSALP) in the presence of the p.Leu299Pro mutation. The simulations indicated alterations in structural flexibility near the mutation site, with potential ramifications for the enzyme's overall stability and function. These dynamic changes may influence the catalytic efficiency of TNSALP, shedding light on the molecular underpinnings of the observed clinical manifestations within the Tunisian family. The clinical presentation of affected individuals highlighted significant phenotypic heterogeneity, underscoring the complex genotype-phenotype correlations in familial Hypophosphatasia. Variability in age of onset, severity of symptoms, and radiographic features was observed, emphasizing the need for a nuanced understanding of the clinical spectrum associated with the p.Leu299Pro mutation. This study advances our understanding of familial Hypophosphatasia by delineating the molecular consequences of the p.Leu299Pro mutation in the ALPL gene. By integrating genetic, structural, and clinical analyses, we provide insights into disease pathogenesis and lay the groundwork for personalized therapeutic strategies tailored to specific genetic profiles. Our findings underscore the importance of comprehensive genetic and clinical evaluation in guiding precision medicine approaches for familial Hypophosphatasia.</p>","PeriodicalId":18816,"journal":{"name":"Molecular Genetics and Genomics","volume":"299 1","pages":"64"},"PeriodicalIF":2.3000,"publicationDate":"2024-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics and Genomics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00438-024-02157-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
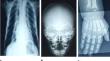
Familial Hypophosphatasia presents a complex diagnostic challenge due to its wide-ranging clinical manifestations and genetic heterogeneity. This study aims to elucidate the molecular underpinnings of familial Hypophosphatasia within a Tunisian family harboring a rare c.896 T > C mutation in the ALPL gene, offering insights into genotype-phenotype correlations and potential therapeutic avenues. The study employs a comprehensive approach, integrating biochemical examination, genetic analysis, structural modeling, and functional insights to unravel the impact of this rare mutation. Genetic investigation revealed the presence of the p.Leu299Pro mutation within the ALPL gene in affected family members. This mutation is strategically positioned in proximity to both the catalytic site and the metal-binding domain, suggesting potential functional consequences. Homology modeling techniques were employed to predict the 3D structure of TNSALP, providing insights into the structural context of the mutation. Our findings suggest that the mutation may induce conformational changes in the vicinity of the catalytic site and metal-binding domain, potentially affecting substrate recognition and catalytic efficiency. Molecular dynamics simulations were instrumental in elucidating the dynamic behavior of the tissue-nonspecific alkaline phosphatase isozyme (TNSALP) in the presence of the p.Leu299Pro mutation. The simulations indicated alterations in structural flexibility near the mutation site, with potential ramifications for the enzyme's overall stability and function. These dynamic changes may influence the catalytic efficiency of TNSALP, shedding light on the molecular underpinnings of the observed clinical manifestations within the Tunisian family. The clinical presentation of affected individuals highlighted significant phenotypic heterogeneity, underscoring the complex genotype-phenotype correlations in familial Hypophosphatasia. Variability in age of onset, severity of symptoms, and radiographic features was observed, emphasizing the need for a nuanced understanding of the clinical spectrum associated with the p.Leu299Pro mutation. This study advances our understanding of familial Hypophosphatasia by delineating the molecular consequences of the p.Leu299Pro mutation in the ALPL gene. By integrating genetic, structural, and clinical analyses, we provide insights into disease pathogenesis and lay the groundwork for personalized therapeutic strategies tailored to specific genetic profiles. Our findings underscore the importance of comprehensive genetic and clinical evaluation in guiding precision medicine approaches for familial Hypophosphatasia.
期刊介绍:
Molecular Genetics and Genomics (MGG) publishes peer-reviewed articles covering all areas of genetics and genomics. Any approach to the study of genes and genomes is considered, be it experimental, theoretical or synthetic. MGG publishes research on all organisms that is of broad interest to those working in the fields of genetics, genomics, biology, medicine and biotechnology.
The journal investigates a broad range of topics, including these from recent issues: mechanisms for extending longevity in a variety of organisms; screening of yeast metal homeostasis genes involved in mitochondrial functions; molecular mapping of cultivar-specific avirulence genes in the rice blast fungus and more.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


