Wenqiang Yang, Kareem E. Abdelfatah, Subrata Kumar Kundu, Biplab Rajbanshi, Gabriel A. Terejanu* and Andreas Heyden*,
{"title":"Machine Learning Accelerated First-Principles Study of the Hydrodeoxygenation of Propanoic Acid","authors":"Wenqiang Yang, Kareem E. Abdelfatah, Subrata Kumar Kundu, Biplab Rajbanshi, Gabriel A. Terejanu* and Andreas Heyden*, ","doi":"10.1021/acscatal.4c01419","DOIUrl":null,"url":null,"abstract":"<p >The complex reaction network of catalytic biomass conversions often involves hundreds of surface intermediates and thousands of reaction steps, greatly hindering the rational design of metal catalysts for these conversions. Here, we present a framework of machine learning (ML)-accelerated first-principles studies for the hydrodeoxygenation (HDO) of propanoic acid over transition metal surfaces. The microkinetic model (MKM) is initially parametrized by ML-predicted energies and iteratively improved by identifying the rate-determining species and steps (RDS), computing their energies by density functional theory (DFT), and reparameterizing the MKM until all the RDS are computed by DFT. The Gaussian process (GP) model performs significantly better than the linear ridge regression model for predicting both the adsorption free energies and transition state free energies. Parameterized with energies from the GP model, only 5–20% of the full reaction network has to be computed by DFT for the MKM to possess DFT-level accuracy for the TOF and dominant reaction pathway. While the linear ridge regression model performs worse than the GP model, its performance is greatly improved when only transition states are predicted by the regression model and adsorption energies are computed by DFT. Overall, we find that a high accuracy in adsorption free energies is more important for a reliable MKM than a high accuracy in TS free energies. Finally, based on the GP model with G<sub>OH</sub> and G<sub>CHCHCO</sub> as catalyst descriptors, we build two-dimensional volcano plots in activity and selectivity that can help design promising alloy catalysts for HDO reactions of organic acids.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":null,"pages":null},"PeriodicalIF":11.3000,"publicationDate":"2024-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.4c01419","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
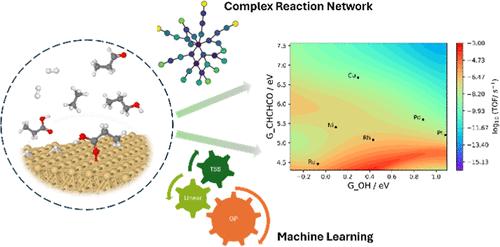
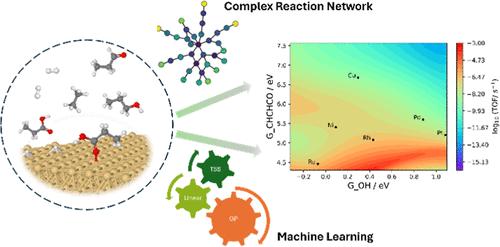
The complex reaction network of catalytic biomass conversions often involves hundreds of surface intermediates and thousands of reaction steps, greatly hindering the rational design of metal catalysts for these conversions. Here, we present a framework of machine learning (ML)-accelerated first-principles studies for the hydrodeoxygenation (HDO) of propanoic acid over transition metal surfaces. The microkinetic model (MKM) is initially parametrized by ML-predicted energies and iteratively improved by identifying the rate-determining species and steps (RDS), computing their energies by density functional theory (DFT), and reparameterizing the MKM until all the RDS are computed by DFT. The Gaussian process (GP) model performs significantly better than the linear ridge regression model for predicting both the adsorption free energies and transition state free energies. Parameterized with energies from the GP model, only 5–20% of the full reaction network has to be computed by DFT for the MKM to possess DFT-level accuracy for the TOF and dominant reaction pathway. While the linear ridge regression model performs worse than the GP model, its performance is greatly improved when only transition states are predicted by the regression model and adsorption energies are computed by DFT. Overall, we find that a high accuracy in adsorption free energies is more important for a reliable MKM than a high accuracy in TS free energies. Finally, based on the GP model with GOH and GCHCHCO as catalyst descriptors, we build two-dimensional volcano plots in activity and selectivity that can help design promising alloy catalysts for HDO reactions of organic acids.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


