The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins
IF 12.9
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Artificial intelligence-driven advances in protein structure prediction in recent years have raised the question: has the protein structure-prediction problem been solved? Here, with a focus on nonglobular proteins, we highlight the many strengths and potential weaknesses of DeepMind’s AlphaFold2 in the context of its biological and therapeutic applications. We summarize the subtleties associated with evaluation of AlphaFold2 model quality and reliability using the predicted local distance difference test (pLDDT) and predicted aligned error (PAE) values. We highlight various classes of proteins that AlphaFold2 can be applied to and the caveats involved. Concrete examples of how AlphaFold2 models can be integrated with experimental data in the form of small-angle X-ray scattering (SAXS), solution NMR, cryo-electron microscopy (cryo-EM) and X-ray diffraction are discussed. Finally, we highlight the need to move beyond structure prediction of rigid, static structural snapshots toward conformational ensembles and alternate biologically relevant states. The overarching theme is that careful consideration is due when using AlphaFold2-generated models to generate testable hypotheses and structural models, rather than treating predicted models as de facto ground truth structures. This Perspective proposes practical guidance to the application of AlphaFold2 for structure prediction of different classes of proteins including rigid globular proteins, intrinsically disordered proteins and alternative conformational states. The use of evaluation metrics to predict reliability of the resulting models and their integration with experimental data are also discussed.
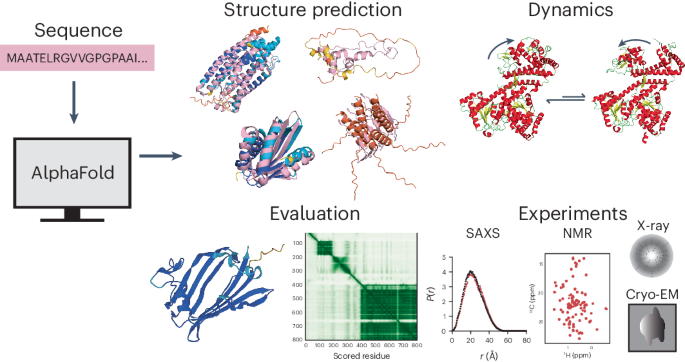
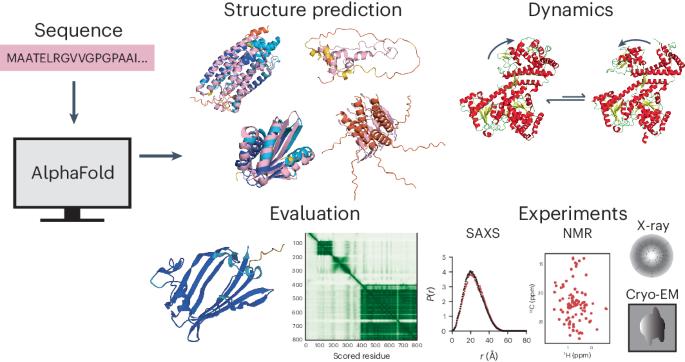
AlphaFold2 在预测刚性球蛋白以外结构方面的能力和缺陷
近年来,人工智能在蛋白质结构预测方面的进步提出了这样一个问题:蛋白质结构预测问题已经解决了吗?在此,我们以非球蛋白为重点,强调 DeepMind 的 AlphaFold2 在生物和治疗应用方面的诸多优势和潜在不足。我们总结了使用预测局部距离差检验(pLDDT)和预测对齐误差(PAE)值评估 AlphaFold2 模型质量和可靠性的相关微妙之处。我们强调了 AlphaFold2 可应用于的各类蛋白质及其注意事项。我们还讨论了 AlphaFold2 模型如何与小角 X 射线散射(SAXS)、溶液 NMR、冷冻电镜(cryo-EM)和 X 射线衍射等形式的实验数据相结合的具体实例。最后,我们强调了超越僵化、静态结构快照的结构预测,转向构象组合和替代生物相关状态的必要性。最重要的主题是,在使用 AlphaFold2 生成的模型来生成可检验的假设和结构模型时,应慎重考虑,而不是将预测的模型视为事实上的基本真理结构。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature chemical biology
生物-生化与分子生物学
CiteScore
23.90
自引率
1.40%
发文量
238
审稿时长
12 months
期刊介绍:
Nature Chemical Biology stands as an esteemed international monthly journal, offering a prominent platform for the chemical biology community to showcase top-tier original research and commentary. Operating at the crossroads of chemistry, biology, and related disciplines, chemical biology utilizes scientific ideas and approaches to comprehend and manipulate biological systems with molecular precision.
The journal embraces contributions from the growing community of chemical biologists, encompassing insights from chemists applying principles and tools to biological inquiries and biologists striving to comprehend and control molecular-level biological processes. We prioritize studies unveiling significant conceptual or practical advancements in areas where chemistry and biology intersect, emphasizing basic research, especially those reporting novel chemical or biological tools and offering profound molecular-level insights into underlying biological mechanisms.
Nature Chemical Biology also welcomes manuscripts describing applied molecular studies at the chemistry-biology interface due to the broad utility of chemical biology approaches in manipulating or engineering biological systems. Irrespective of scientific focus, we actively seek submissions that creatively blend chemistry and biology, particularly those providing substantial conceptual or methodological breakthroughs with the potential to open innovative research avenues. The journal maintains a robust and impartial review process, emphasizing thorough chemical and biological characterization.
文献相关原料
| 公司名称 | 产品信息 | 采购帮参考价格 |
|---|
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


