A.L. Okana-Lomanga , G. Dimitri Ngantso , B.R. Malonda-Boungou , A.T. Raji , B. M'Passi-Mabiala
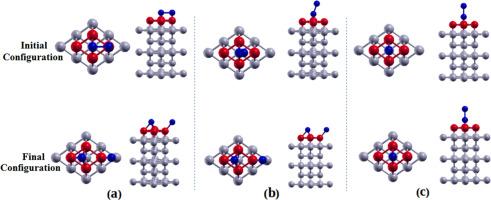
{"title":"Adsorption of dihalogen molecules X2 (X = F, Cl, Br and I) on the Fe/W(110) substrate","authors":"A.L. Okana-Lomanga , G. Dimitri Ngantso , B.R. Malonda-Boungou , A.T. Raji , B. M'Passi-Mabiala","doi":"10.1016/j.susc.2024.122536","DOIUrl":null,"url":null,"abstract":"<div><p>We report on spin-polarized density-functional theory study of adsorption of dihalogen molecules X<sub>2</sub> (<em>X</em> = <em>F</em>, Cl, Br and I) on the Fe/W(110) substrate, i.e., X<sub>2</sub>/Fe/W(110) systems. We considered different molecular orientations and adsorption sites of the halogens and obtained their corresponding ground-state structures. We obtained initial molecular orientation (IMO) and initial adsorption site (IAS), i.e., IMO-IAS combinations that give the minimum energy configurations for each of the X<sub>2</sub>/Fe/W(110) systems. Our results shows that all the molecules studied in this work are chemisorbed on the Fe surface. Also, the halogen atoms may be adsorbed dissociatively on the <em>hollow</em> sites in such a way that an X<sub>2</sub> separates into two X atoms with each of the atoms located at two nearby <em>hollow</em> sites. Similarly, we found IMO-IAS combinations which resulted in a non-dissociative adsorption. In the latter, the pre-relaxed IMO-IAS is maintained even after the structural relaxation. The most stable configuration for the X<sub>2</sub> dihalogen molecule in this case is either the <em>top</em> or <em>bridge</em> site while the halogen is in perpendicular orientation to the Fe surface. We conclude therefore that, the final relaxed configurations of the X<sub>2</sub> halogen depends on the IMO through which is deposited on the Fe/W(110) substrate. The trend in the adsorption energy E<sub>A</sub> for the most stable configurations for the dissociative adsorption is <em>E<sub>A</sub></em> (F) > <em>E<sub>A</sub></em> (Cl) > <em>E<sub>A</sub></em> (Br) > <em>E<sub>A</sub></em> (I). The trend of <em>E</em><sub>A</sub> for non-dissociative adsorption is similar to that of dissociative adsorption, however, the latter is the more energetically favorable. Electronic structure calculations show hybridization between the <em>p</em> and <em>d</em> orbitals of X and Fe atoms respectively. Furthermore, we have found antiferromagnetic coupling between the interfacial W atoms and the Fe overlayer atoms while ferromagnetic coupling is found between the halogens and the Fe atoms. Our work represents a detailed study of adsorption properties of highly reactive halogens in contact with the Fe/W(100) surface.</p></div>","PeriodicalId":22100,"journal":{"name":"Surface Science","volume":"748 ","pages":"Article 122536"},"PeriodicalIF":2.1000,"publicationDate":"2024-06-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Surface Science","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0039602824000876","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
We report on spin-polarized density-functional theory study of adsorption of dihalogen molecules X2 (X = F, Cl, Br and I) on the Fe/W(110) substrate, i.e., X2/Fe/W(110) systems. We considered different molecular orientations and adsorption sites of the halogens and obtained their corresponding ground-state structures. We obtained initial molecular orientation (IMO) and initial adsorption site (IAS), i.e., IMO-IAS combinations that give the minimum energy configurations for each of the X2/Fe/W(110) systems. Our results shows that all the molecules studied in this work are chemisorbed on the Fe surface. Also, the halogen atoms may be adsorbed dissociatively on the hollow sites in such a way that an X2 separates into two X atoms with each of the atoms located at two nearby hollow sites. Similarly, we found IMO-IAS combinations which resulted in a non-dissociative adsorption. In the latter, the pre-relaxed IMO-IAS is maintained even after the structural relaxation. The most stable configuration for the X2 dihalogen molecule in this case is either the top or bridge site while the halogen is in perpendicular orientation to the Fe surface. We conclude therefore that, the final relaxed configurations of the X2 halogen depends on the IMO through which is deposited on the Fe/W(110) substrate. The trend in the adsorption energy EA for the most stable configurations for the dissociative adsorption is EA (F) > EA (Cl) > EA (Br) > EA (I). The trend of EA for non-dissociative adsorption is similar to that of dissociative adsorption, however, the latter is the more energetically favorable. Electronic structure calculations show hybridization between the p and d orbitals of X and Fe atoms respectively. Furthermore, we have found antiferromagnetic coupling between the interfacial W atoms and the Fe overlayer atoms while ferromagnetic coupling is found between the halogens and the Fe atoms. Our work represents a detailed study of adsorption properties of highly reactive halogens in contact with the Fe/W(100) surface.
我们报告了二卤素分子 X2(X = F、Cl、Br 和 I)在 Fe/W(110)基底(即 X2/Fe/W(110) 系统)上吸附的自旋极化密度泛函理论研究。我们考虑了卤素的不同分子取向和吸附位点,并得到了其相应的基态结构。我们得到了初始分子取向(IMO)和初始吸附位点(IAS),即 IMO-IAS 组合,这些组合给出了每个 X2/Fe/W(110) 系统的最小能量构型。我们的研究结果表明,这项工作中研究的所有分子都在铁表面发生了化学吸附。此外,卤素原子可能以离解的方式吸附在空心位点上,从而使一个 X2 分离成两个 X 原子,每个原子都位于附近的两个空心位点上。同样,我们发现 IMO-IAS 组合也会导致非离解吸附。在后者中,即使在结构松弛后,也能保持预先松弛的 IMO-IAS 结构。在这种情况下,X2 二卤素分子最稳定的构型是顶部位点或桥位点,而卤素则与铁表面垂直。因此我们得出结论,X2 卤素的最终弛豫构型取决于沉积在 Fe/W(110)基底上的 IMO。解离吸附的最稳定构型的吸附能 EA 的趋势是 EA (F) > EA (Cl) > EA (Br) > EA (I)。非解离吸附的 EA 变化趋势与解离吸附相似,但后者在能量上更为有利。电子结构计算显示,X 原子和 Fe 原子的 p 和 d 轨道之间分别存在杂化现象。此外,我们还发现了界面 W 原子和铁覆盖层原子之间的反铁磁耦合,而卤素和铁原子之间则存在铁磁耦合。我们的工作是对与 Fe/W(100)表面接触的高活性卤素吸附特性的详细研究。
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


