{"title":"Alternate recognition by dengue protease: Proteolytic and binding assays provide functional evidence beyond an induced-fit.","authors":"Mira A M Behnam, Christian D Klein","doi":"10.1016/j.biochi.2024.06.002","DOIUrl":null,"url":null,"abstract":"<p><p>Proteases are key enzymes in viral replication, and interfering with these targets is the basis for therapeutic interventions. We previously introduced a hypothesis about conformational selection in the protease of dengue virus and related flaviviruses, based on conformational plasticity noted in X-ray structures. The present work presents the first functional evidence for alternate recognition by the dengue protease, in a mechanism based primarily on conformational selection rather than induced-fit. Recognition of distinct substrates and inhibitors in proteolytic and binding assays varies to a different extent, depending on factors reported to influence the protease structure. The pH, salinity, buffer type, and temperature cause a change in binding, proteolysis, or inhibition behavior. Using representative inhibitors with distinct structural scaffolds, we identify two contrasting binding profiles to dengue protease. Noticeable effects are observed in the binding assay upon inclusion of a non-ionic detergent in comparison to the proteolytic assay. The findings highlight the impact of the selection of testing conditions on the observed ligand affinity or inhibitory potency. From a broader scope, the dengue protease presents an example, where the induced-fit paradigm appears insufficient to explain binding events with the biological target. Furthermore, this protein reveals the complexity of comparing or combining biochemical assay data obtained under different conditions. This can be particularly critical for artificial intelligence (AI) approaches in drug discovery that rely on large datasets of compounds activity, compiled from different sources using non-identical testing procedures. In such cases, mismatched results will compromise the model quality and its predictive power.</p>","PeriodicalId":93898,"journal":{"name":"Biochimie","volume":" ","pages":"15-27"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochimie","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.biochi.2024.06.002","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/6/11 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
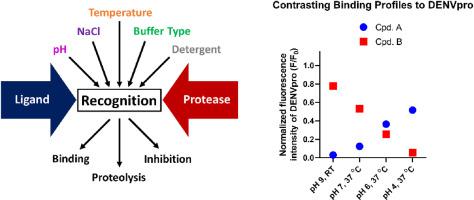
Proteases are key enzymes in viral replication, and interfering with these targets is the basis for therapeutic interventions. We previously introduced a hypothesis about conformational selection in the protease of dengue virus and related flaviviruses, based on conformational plasticity noted in X-ray structures. The present work presents the first functional evidence for alternate recognition by the dengue protease, in a mechanism based primarily on conformational selection rather than induced-fit. Recognition of distinct substrates and inhibitors in proteolytic and binding assays varies to a different extent, depending on factors reported to influence the protease structure. The pH, salinity, buffer type, and temperature cause a change in binding, proteolysis, or inhibition behavior. Using representative inhibitors with distinct structural scaffolds, we identify two contrasting binding profiles to dengue protease. Noticeable effects are observed in the binding assay upon inclusion of a non-ionic detergent in comparison to the proteolytic assay. The findings highlight the impact of the selection of testing conditions on the observed ligand affinity or inhibitory potency. From a broader scope, the dengue protease presents an example, where the induced-fit paradigm appears insufficient to explain binding events with the biological target. Furthermore, this protein reveals the complexity of comparing or combining biochemical assay data obtained under different conditions. This can be particularly critical for artificial intelligence (AI) approaches in drug discovery that rely on large datasets of compounds activity, compiled from different sources using non-identical testing procedures. In such cases, mismatched results will compromise the model quality and its predictive power.
蛋白酶是病毒复制的关键酶,干扰这些靶点是治疗干预的基础。此前,我们根据 X 射线结构中的构象可塑性,提出了登革病毒和相关黄病毒蛋白酶构象选择的假说。本研究首次提出了登革病毒蛋白酶交替识别的功能证据,其机制主要基于构象选择而非诱导拟合。在蛋白水解和结合试验中,对不同底物和抑制剂的识别会有不同程度的变化,这取决于据报道会影响蛋白酶结构的因素。pH 值、盐度、缓冲液类型和温度会导致结合、蛋白水解或抑制行为发生变化。利用具有不同结构支架的代表性抑制剂,我们发现了登革热蛋白酶的两种截然不同的结合情况。与蛋白水解试验相比,在结合试验中加入非离子去垢剂会产生明显的影响。这些发现凸显了测试条件的选择对观察到的配体亲和力或抑制效力的影响。从更广的范围来看,登革热蛋白酶是一个例子,在这个例子中,诱导拟合模式似乎不足以解释与生物靶标的结合事件。此外,这种蛋白质还揭示了比较或结合在不同条件下获得的生化检测数据的复杂性。这对于药物发现中的人工智能(AI)方法尤为重要,因为这些方法依赖于大量的化合物活性数据集,而这些数据集是使用非相同的测试程序从不同来源收集的。在这种情况下,不匹配的结果会影响模型的质量和预测能力。

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


