Clinical and molecular characteristics of Korean patients with Kabuki syndrome
IF 2.6
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
Abstract
Kabuki syndrome (KS) is a rare disorder characterized by typical facial features, skeletal anomalies, fetal fingertip pad persistence, postnatal growth retardation, and intellectual disabilities. Heterozygous variants of the KMT2D and KDM6A genes are major genetic causes of KS. This study aimed to report the clinical and genetic characteristics of KS. This study included 28 Korean patients (14 boys and 14 girls) with KS through molecular genetic testing, including direct Sanger sequencing, whole-exome sequencing, or whole-genome sequencing. The median age at clinical diagnosis was 18.5 months (IQR 7–58 months), and the median follow-up duration was 80.5 months (IQR 48–112 months). Molecular genetic testing identified different pathogenic variants of the KMT2D (n = 23) and KDM6A (n = 3) genes, including 15 novel variants. Patients showed typical facial features (100%), such as long palpebral fissure and eversion of the lower eyelid; intellectual disability/developmental delay (96%); short stature (79%); and congenital cardiac anomalies (75%). Although 71% experienced failure to thrive in infancy, 54% of patients showed a tendency toward overweight/obesity in early childhood. Patients with KDM6A variants demonstrated severe genotype-phenotype correlation. This study enhances the understanding of the clinical and genetic characteristics of KS.
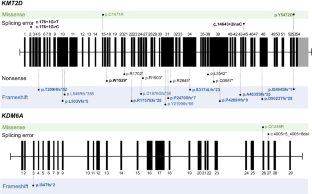
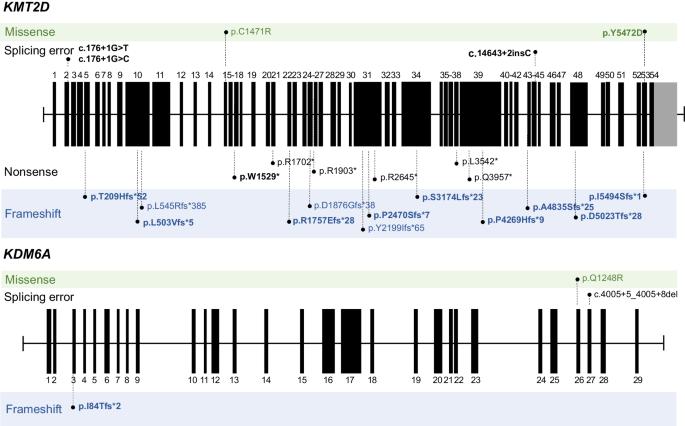
韩国歌舞伎综合征患者的临床和分子特征。
简介歌舞伎综合征(KS)是一种罕见的疾病,具有典型的面部特征、骨骼异常、胎儿指垫持续存在、出生后生长迟缓和智力障碍等特征。KMT2D和KDM6A基因的杂合子变异是KS的主要遗传病因。本研究旨在报告 KS 的临床和遗传特征:本研究通过分子遗传学检测,包括直接桑格测序、全外显子组测序或全基因组测序,纳入了 28 名韩国 KS 患者(14 名男孩和 14 名女孩):临床诊断时的中位年龄为 18.5 个月(IQR 7-58 个月),中位随访时间为 80.5 个月(IQR 48-112 个月)。分子基因检测发现了KMT2D(23个)和KDM6A(3个)基因的不同致病变体,其中包括15个新型变体。患者表现出典型的面部特征(100%),如长睑裂和下眼睑外翻;智力残疾/发育迟缓(96%);身材矮小(79%);先天性心脏异常(75%)。虽然71%的患者在婴儿期无法茁壮成长,但54%的患者在幼儿期表现出超重/肥胖倾向。KDM6A变体患者表现出严重的基因型与表型相关性:这项研究加深了人们对 KS 临床和遗传特征的了解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


