Honggang Zhao, Ximing Guo, Wenlu Wang, Zhenwei Wang, Paul Rawson, Ami Wilbur, Matthew Hare
{"title":"Consequences of domestication in eastern oyster: Insights from whole genomic analyses","authors":"Honggang Zhao, Ximing Guo, Wenlu Wang, Zhenwei Wang, Paul Rawson, Ami Wilbur, Matthew Hare","doi":"10.1111/eva.13710","DOIUrl":null,"url":null,"abstract":"<p>Selective breeding for production traits has yielded relatively rapid successes with high-fecundity aquaculture species. Discovering the genetic changes associated with selection is an important goal for understanding adaptation and can also facilitate better predictions about the likely fitness of selected strains if they escape aquaculture farms. Here, we hypothesize domestication as a genetic change induced by inadvertent selection in culture. Our premise is that standardized culture protocols generate parallel domestication effects across independent strains. Using eastern oyster as a model and a newly developed 600K SNP array, this study tested for parallel domestication effects in multiple independent selection lines compared with their progenitor wild populations. A single contrast was made between pooled selected strains (1–17 generations in culture) and all wild progenitor samples combined. Population structure analysis indicated rank order levels of differentiation as [wild − wild] < [wild − cultured] < [cultured − cultured]. A genome scan for parallel adaptation to the captive environment applied two methodologically distinct outlier tests to the wild versus selected strain contrast and identified a total of 1174 candidate SNPs. Contrasting wild versus selected strains revealed the early evolutionary consequences of domestication in terms of genomic differentiation, standing genetic diversity, effective population size, relatedness, runs of homozygosity profiles, and genome-wide linkage disequilibrium patterns. Random Forest was used to identify 37 outlier SNPs that had the greatest discriminatory power between bulked wild and selected oysters. The outlier SNPs were in genes enriched for cytoskeletal functions, hinting at possible traits under inadvertent selection during larval culture or pediveliger setting at high density. This study documents rapid genomic changes stemming from hatchery-based cultivation of eastern oysters, identifies candidate loci responding to domestication in parallel among independent aquaculture strains, and provides potentially useful genomic resources for monitoring interbreeding between farm and wild oysters.</p>","PeriodicalId":168,"journal":{"name":"Evolutionary Applications","volume":"17 6","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2024-05-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/eva.13710","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Evolutionary Applications","FirstCategoryId":"99","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/eva.13710","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
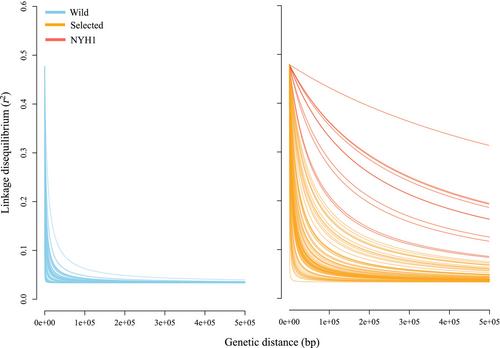
Selective breeding for production traits has yielded relatively rapid successes with high-fecundity aquaculture species. Discovering the genetic changes associated with selection is an important goal for understanding adaptation and can also facilitate better predictions about the likely fitness of selected strains if they escape aquaculture farms. Here, we hypothesize domestication as a genetic change induced by inadvertent selection in culture. Our premise is that standardized culture protocols generate parallel domestication effects across independent strains. Using eastern oyster as a model and a newly developed 600K SNP array, this study tested for parallel domestication effects in multiple independent selection lines compared with their progenitor wild populations. A single contrast was made between pooled selected strains (1–17 generations in culture) and all wild progenitor samples combined. Population structure analysis indicated rank order levels of differentiation as [wild − wild] < [wild − cultured] < [cultured − cultured]. A genome scan for parallel adaptation to the captive environment applied two methodologically distinct outlier tests to the wild versus selected strain contrast and identified a total of 1174 candidate SNPs. Contrasting wild versus selected strains revealed the early evolutionary consequences of domestication in terms of genomic differentiation, standing genetic diversity, effective population size, relatedness, runs of homozygosity profiles, and genome-wide linkage disequilibrium patterns. Random Forest was used to identify 37 outlier SNPs that had the greatest discriminatory power between bulked wild and selected oysters. The outlier SNPs were in genes enriched for cytoskeletal functions, hinting at possible traits under inadvertent selection during larval culture or pediveliger setting at high density. This study documents rapid genomic changes stemming from hatchery-based cultivation of eastern oysters, identifies candidate loci responding to domestication in parallel among independent aquaculture strains, and provides potentially useful genomic resources for monitoring interbreeding between farm and wild oysters.
期刊介绍:
Evolutionary Applications is a fully peer reviewed open access journal. It publishes papers that utilize concepts from evolutionary biology to address biological questions of health, social and economic relevance. Papers are expected to employ evolutionary concepts or methods to make contributions to areas such as (but not limited to): medicine, agriculture, forestry, exploitation and management (fisheries and wildlife), aquaculture, conservation biology, environmental sciences (including climate change and invasion biology), microbiology, and toxicology. All taxonomic groups are covered from microbes, fungi, plants and animals. In order to better serve the community, we also now strongly encourage submissions of papers making use of modern molecular and genetic methods (population and functional genomics, transcriptomics, proteomics, epigenetics, quantitative genetics, association and linkage mapping) to address important questions in any of these disciplines and in an applied evolutionary framework. Theoretical, empirical, synthesis or perspective papers are welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


