Repurposing a plant peptide cyclase for targeted lysine acylation
IF 19.2
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
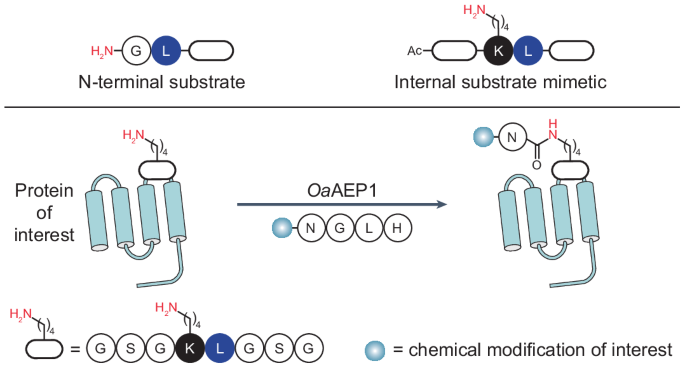
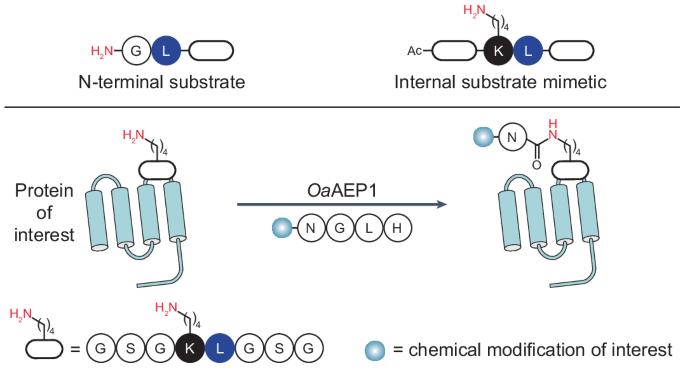
Transpeptidases are powerful tools for protein engineering but are largely restricted to acting at protein backbone termini. Alternative enzymatic approaches for internal protein labelling require bulky recognition motifs or non-proteinogenic reaction partners, potentially restricting which proteins can be modified or the types of modification that can be installed. Here we report a strategy for labelling lysine side chain ε-amines by repurposing an engineered asparaginyl ligase, which naturally catalyses peptide head-to-tail cyclization, for versatile isopeptide ligations that are compatible with peptidic substrates. We find that internal lysines with an adjacent leucine residue mimic the conventional N-terminal glycine–leucine substrate. This dipeptide motif enables efficient intra- or intermolecular ligation through internal lysine side chains, minimally leaving an asparagine C-terminally linked to the lysine side chain via an isopeptide bond. The versatility of this approach is demonstrated by the chemoenzymatic synthesis of peptides with non-native C terminus-to-side chain topology and the conjugation of chemically modified peptides to recombinant proteins. Plant asparaginyl endopeptidases that function preferentially as transpeptidases naturally catalyse the head-to-tail cyclization of plant peptides. Using substrate mimicry and reaction optimization, their function has now been repurposed to catalyse intermolecular isopeptide bond formation on diverse peptide and protein substrates.
将植物肽环化酶重新用于靶向赖氨酸酰化
反肽酶是蛋白质工程的强大工具,但在很大程度上仅限于作用于蛋白质骨架末端。用于蛋白质内部标记的其他酶法需要大体积识别基团或非蛋白源反应伙伴,这可能会限制可修饰的蛋白质或可安装的修饰类型。在这里,我们报告了一种标记赖氨酸侧链ε-胺的策略,它将一种天然催化肽头尾环化的天冬酰胺连接酶重新用于与肽基质兼容的多功能异肽连接。我们发现,内部赖氨酸与相邻的亮氨酸残基可模拟传统的 N 端甘氨酸-亮氨酸底物。这种二肽结构可通过内部赖氨酸侧链实现高效的分子内或分子间连接,并通过等肽键将天冬酰胺 C 端与赖氨酸侧链连接在一起。通过化学酶法合成具有非原生 C 端到侧链拓扑结构的肽,以及将化学修饰的肽与重组蛋白连接,证明了这种方法的多功能性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature chemistry
化学-化学综合
CiteScore
29.60
自引率
1.40%
发文量
226
审稿时长
1.7 months
期刊介绍:
Nature Chemistry is a monthly journal that publishes groundbreaking and significant research in all areas of chemistry. It covers traditional subjects such as analytical, inorganic, organic, and physical chemistry, as well as a wide range of other topics including catalysis, computational and theoretical chemistry, and environmental chemistry.
The journal also features interdisciplinary research at the interface of chemistry with biology, materials science, nanotechnology, and physics. Manuscripts detailing such multidisciplinary work are encouraged, as long as the central theme pertains to chemistry.
Aside from primary research, Nature Chemistry publishes review articles, news and views, research highlights from other journals, commentaries, book reviews, correspondence, and analysis of the broader chemical landscape. It also addresses crucial issues related to education, funding, policy, intellectual property, and the societal impact of chemistry.
Nature Chemistry is dedicated to ensuring the highest standards of original research through a fair and rigorous review process. It offers authors maximum visibility for their papers, access to a broad readership, exceptional copy editing and production standards, rapid publication, and independence from academic societies and other vested interests.
Overall, Nature Chemistry aims to be the authoritative voice of the global chemical community.
文献相关原料
| 公司名称 | 产品信息 | 采购帮参考价格 |
|---|
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


