An MSstats workflow for detecting differentially abundant proteins in large-scale data-independent acquisition mass spectrometry experiments with FragPipe processing
IF 13.1
1区 生物学
Q1 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Technological advances in mass spectrometry and proteomics have made it possible to perform larger-scale and more-complex experiments. The volume and complexity of the resulting data create major challenges for downstream analysis. In particular, next-generation data-independent acquisition (DIA) experiments enable wider proteome coverage than more traditional targeted approaches but require computational workflows that can manage much larger datasets and identify peptide sequences from complex and overlapping spectral features. Data-processing tools such as FragPipe, DIA-NN and Spectronaut have undergone substantial improvements to process spectral features in a reasonable time. Statistical analysis tools are needed to draw meaningful comparisons between experimental samples, but these tools were also originally designed with smaller datasets in mind. This protocol describes an updated version of MSstats that has been adapted to be compatible with large-scale DIA experiments. A very large DIA experiment, processed with FragPipe, is used as an example to demonstrate different MSstats workflows. The choice of workflow depends on the user’s computational resources. For datasets that are too large to fit into a standard computer’s memory, we demonstrate the use of MSstatsBig, a companion R package to MSstats. The protocol also highlights key decisions that have a major effect on both the results and the processing time of the analysis. The MSstats processing can be expected to take 1–3 h depending on the usage of MSstatsBig. The protocol can be run in the point-and-click graphical user interface MSstatsShiny or implemented with minimal coding expertise in R. MSstats is used for the statistical analysis of proteomics data from DIA mass spectrometry experiments. This protocol describes workflows using (i) the MSstatsShiny graphical user interface and (ii) the R packages MSstats or MSstatsBig (for larger datasets).
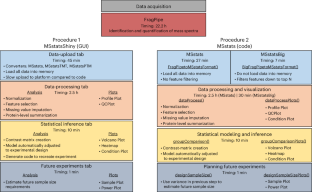
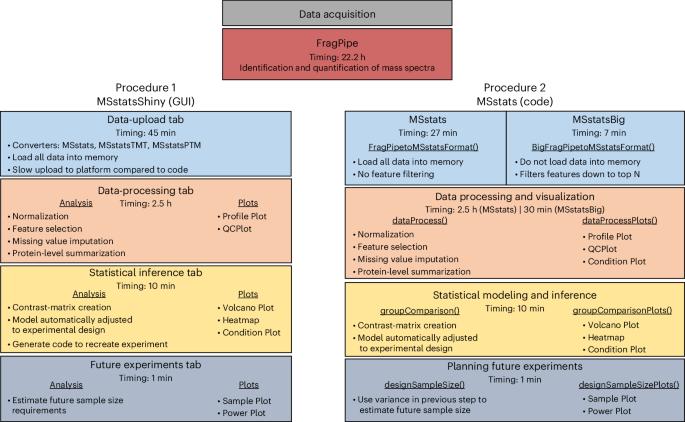
利用 FragPipe 处理在大规模数据独立采集质谱实验中检测差异丰富蛋白质的 MSstats 工作流程。
质谱仪和蛋白质组学技术的进步使得进行更大规模、更复杂的实验成为可能。由此产生的数据量和复杂性给下游分析带来了重大挑战。特别是与传统的靶向方法相比,下一代数据独立获取(DIA)实验能够实现更广泛的蛋白质组覆盖,但需要能够管理大得多的数据集并从复杂和重叠的光谱特征中识别肽序列的计算工作流。数据处理工具(如 FragPipe、DIA-NN 和 Spectronaut)在合理时间内处理光谱特征方面有了很大改进。需要使用统计分析工具对实验样本进行有意义的比较,但这些工具最初也是针对较小的数据集设计的。本协议描述了 MSstats 的更新版本,该版本已进行了调整,以兼容大规模 DIA 实验。以一个用 FragPipe 处理的超大型 DIA 实验为例,演示了不同的 MSstats 工作流程。工作流程的选择取决于用户的计算资源。如果数据集过大,标准计算机内存无法容纳,我们将演示 MSstatsBig(MSstats 的配套 R 软件包)的使用。该协议还强调了对分析结果和处理时间有重大影响的关键决策。根据 MSstatsBig 的使用情况,MSstats 处理预计需要 1-3 小时。该协议可在点选式图形用户界面 MSstatsShiny 中运行,也可在 R 语言中以最低限度的专业编码技术实现。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Protocols
生物-生化研究方法
CiteScore
29.10
自引率
0.70%
发文量
128
审稿时长
4 months
期刊介绍:
Nature Protocols focuses on publishing protocols used to address significant biological and biomedical science research questions, including methods grounded in physics and chemistry with practical applications to biological problems. The journal caters to a primary audience of research scientists and, as such, exclusively publishes protocols with research applications. Protocols primarily aimed at influencing patient management and treatment decisions are not featured.
The specific techniques covered encompass a wide range, including but not limited to: Biochemistry, Cell biology, Cell culture, Chemical modification, Computational biology, Developmental biology, Epigenomics, Genetic analysis, Genetic modification, Genomics, Imaging, Immunology, Isolation, purification, and separation, Lipidomics, Metabolomics, Microbiology, Model organisms, Nanotechnology, Neuroscience, Nucleic-acid-based molecular biology, Pharmacology, Plant biology, Protein analysis, Proteomics, Spectroscopy, Structural biology, Synthetic chemistry, Tissue culture, Toxicology, and Virology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


