Sakina Bombaywala , Abhay Bajaj , Nishant A. Dafale
{"title":"Meta-analysis of wastewater microbiome for antibiotic resistance profiling","authors":"Sakina Bombaywala , Abhay Bajaj , Nishant A. Dafale","doi":"10.1016/j.mimet.2024.106953","DOIUrl":null,"url":null,"abstract":"<div><p>The microbial composition and stress molecules are main drivers influencing the development and spread of antibiotic resistance bacteria (ARBs) and genes (ARGs) in the environment. A reliable and rapid method for identifying associations between microbiome composition and resistome remains challenging. In the present study, secondary metagenome data of sewage and hospital wastewaters were assessed for differential taxonomic and ARG profiling. Subsequently, Random Forest (RF)-based ML models were used to predict ARG profiles based on taxonomic composition and model validation on hospital wastewaters. Total ARG abundance was significantly higher in hospital wastewaters (15 ppm) than sewage (5 ppm), while the resistance towards methicillin, carbapenem, and fluoroquinolone were predominant. Although, <em>Pseudomonas</em> constituted major fraction, <em>Streptomyces, Enterobacter</em>, and <em>Klebsiella</em> were characteristic of hospital wastewaters. Prediction modeling showed that the relative abundance of pathogenic genera <em>Escherichia, Vibrio,</em> and <em>Pseudomonas</em> contributed most towards variations in total ARG count. Moreover, the model was able to identify host-specific patterns for contributing taxa and related ARGs with >90% accuracy in predicting the ARG subtype abundance. More than >80% accuracy was obtained for hospital wastewaters, demonstrating that the model can be validly extrapolated to different types of wastewater systems. Findings from the study showed that the ML approach could identify ARG profile based on bacterial composition including 16S rDNA amplicon data, and can serve as a viable alternative to metagenomic binning for identification of potential hosts of ARGs. Overall, this study demonstrates the promising application of ML techniques for predicting the spread of ARGs and provides guidance for early warning of ARBs emergence.</p></div>","PeriodicalId":16409,"journal":{"name":"Journal of microbiological methods","volume":"223 ","pages":"Article 106953"},"PeriodicalIF":1.7000,"publicationDate":"2024-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of microbiological methods","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0167701224000654","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
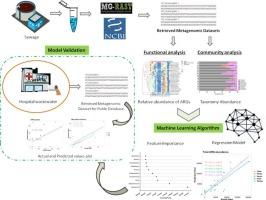
The microbial composition and stress molecules are main drivers influencing the development and spread of antibiotic resistance bacteria (ARBs) and genes (ARGs) in the environment. A reliable and rapid method for identifying associations between microbiome composition and resistome remains challenging. In the present study, secondary metagenome data of sewage and hospital wastewaters were assessed for differential taxonomic and ARG profiling. Subsequently, Random Forest (RF)-based ML models were used to predict ARG profiles based on taxonomic composition and model validation on hospital wastewaters. Total ARG abundance was significantly higher in hospital wastewaters (15 ppm) than sewage (5 ppm), while the resistance towards methicillin, carbapenem, and fluoroquinolone were predominant. Although, Pseudomonas constituted major fraction, Streptomyces, Enterobacter, and Klebsiella were characteristic of hospital wastewaters. Prediction modeling showed that the relative abundance of pathogenic genera Escherichia, Vibrio, and Pseudomonas contributed most towards variations in total ARG count. Moreover, the model was able to identify host-specific patterns for contributing taxa and related ARGs with >90% accuracy in predicting the ARG subtype abundance. More than >80% accuracy was obtained for hospital wastewaters, demonstrating that the model can be validly extrapolated to different types of wastewater systems. Findings from the study showed that the ML approach could identify ARG profile based on bacterial composition including 16S rDNA amplicon data, and can serve as a viable alternative to metagenomic binning for identification of potential hosts of ARGs. Overall, this study demonstrates the promising application of ML techniques for predicting the spread of ARGs and provides guidance for early warning of ARBs emergence.
期刊介绍:
The Journal of Microbiological Methods publishes scholarly and original articles, notes and review articles. These articles must include novel and/or state-of-the-art methods, or significant improvements to existing methods. Novel and innovative applications of current methods that are validated and useful will also be published. JMM strives for scholarship, innovation and excellence. This demands scientific rigour, the best available methods and technologies, correctly replicated experiments/tests, the inclusion of proper controls, calibrations, and the correct statistical analysis. The presentation of the data must support the interpretation of the method/approach.
All aspects of microbiology are covered, except virology. These include agricultural microbiology, applied and environmental microbiology, bioassays, bioinformatics, biotechnology, biochemical microbiology, clinical microbiology, diagnostics, food monitoring and quality control microbiology, microbial genetics and genomics, geomicrobiology, microbiome methods regardless of habitat, high through-put sequencing methods and analysis, microbial pathogenesis and host responses, metabolomics, metagenomics, metaproteomics, microbial ecology and diversity, microbial physiology, microbial ultra-structure, microscopic and imaging methods, molecular microbiology, mycology, novel mathematical microbiology and modelling, parasitology, plant-microbe interactions, protein markers/profiles, proteomics, pyrosequencing, public health microbiology, radioisotopes applied to microbiology, robotics applied to microbiological methods,rumen microbiology, microbiological methods for space missions and extreme environments, sampling methods and samplers, soil and sediment microbiology, transcriptomics, veterinary microbiology, sero-diagnostics and typing/identification.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


