Clara Gontijo Camelo, Cristiane de Araujo Martins Moreno, Mariana da Cunha Artilheiro, Alulin Tácio Quadros Monteiro Fonseca, Juliana Gurgel Gianetti, André Vinícius Barbosa, Karina Carvalho Donis, Jonas Alex Morales Saute, André Pessoa, Hélio Van der Linden Jr, Ana Rita Alcântara Gonçalves, Leslie Domenici Kulikowski, Fernando Kok, Edmar Zanoteli
{"title":"Genetic profile of Brazilian patients with LAMA2-related dystrophies","authors":"Clara Gontijo Camelo, Cristiane de Araujo Martins Moreno, Mariana da Cunha Artilheiro, Alulin Tácio Quadros Monteiro Fonseca, Juliana Gurgel Gianetti, André Vinícius Barbosa, Karina Carvalho Donis, Jonas Alex Morales Saute, André Pessoa, Hélio Van der Linden Jr, Ana Rita Alcântara Gonçalves, Leslie Domenici Kulikowski, Fernando Kok, Edmar Zanoteli","doi":"10.1111/cge.14538","DOIUrl":null,"url":null,"abstract":"<p>LAMA2-related dystrophies (LAMA2-RD) constitute a rare neuromuscular disorder with a broad spectrum of phenotypic severity. Our understanding of the genotype–phenotype correlations in this condition remains incomplete, and reliable clinical data for clinical trial readiness is limited. In this retrospective study, we reviewed the genetic data and medical records of 114 LAMA2-RD patients enrolled at seven research centers in Brazil. We identified 58 different pathogenic variants, including 21 novel ones. Six variants were more prevalent and were present in 81.5% of the patients. Notably, the c.1255del, c.2049_2050del, c.3976 C>T, c.5234+1G>A, and c.4739dup variants were found in patients unable to walk and without cortical malformation. In contrast, the c.2461A>C variant was present in patients who could walk unassisted. Among ambulatory patients, missense variants were more prevalent (<i>p</i> < 0.0001). Although no specific hotspot regions existed in the <i>LAMA2</i>, 51% of point mutations were in the LN domain, and 88% of the missense variants were found within this domain. Functional analysis was performed in one intronic variant (c.4960-17C>A) and revealed an out-of-frame transcript, indicating that the variant creates a cryptic splicing site (AG). Our study has shed light on crucial phenotype–genotype correlations and provided valuable insights, particularly regarding the Latin American population.</p>","PeriodicalId":10354,"journal":{"name":"Clinical Genetics","volume":"106 3","pages":"305-314"},"PeriodicalIF":2.9000,"publicationDate":"2024-05-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Genetics","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/cge.14538","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
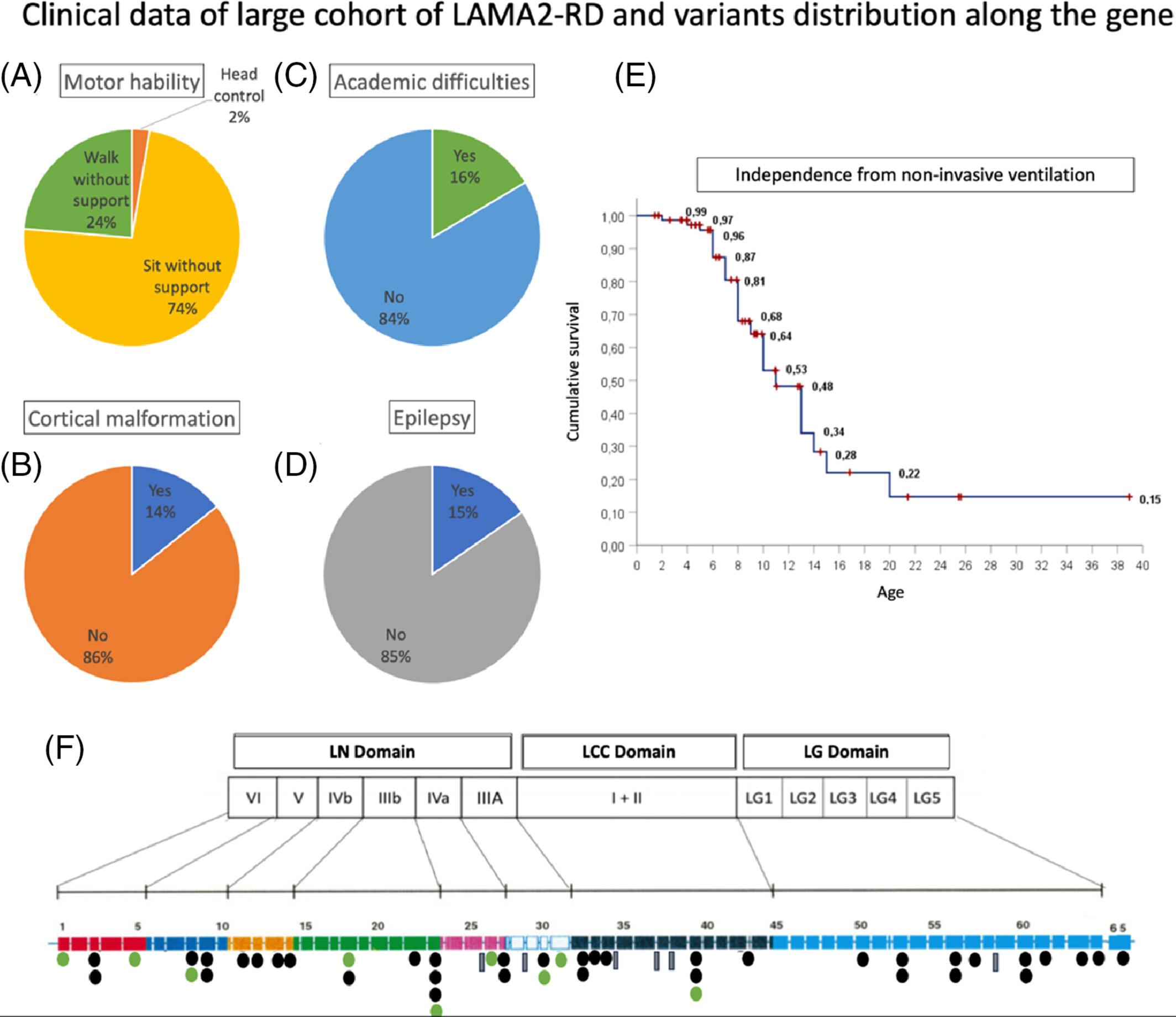
LAMA2-related dystrophies (LAMA2-RD) constitute a rare neuromuscular disorder with a broad spectrum of phenotypic severity. Our understanding of the genotype–phenotype correlations in this condition remains incomplete, and reliable clinical data for clinical trial readiness is limited. In this retrospective study, we reviewed the genetic data and medical records of 114 LAMA2-RD patients enrolled at seven research centers in Brazil. We identified 58 different pathogenic variants, including 21 novel ones. Six variants were more prevalent and were present in 81.5% of the patients. Notably, the c.1255del, c.2049_2050del, c.3976 C>T, c.5234+1G>A, and c.4739dup variants were found in patients unable to walk and without cortical malformation. In contrast, the c.2461A>C variant was present in patients who could walk unassisted. Among ambulatory patients, missense variants were more prevalent (p < 0.0001). Although no specific hotspot regions existed in the LAMA2, 51% of point mutations were in the LN domain, and 88% of the missense variants were found within this domain. Functional analysis was performed in one intronic variant (c.4960-17C>A) and revealed an out-of-frame transcript, indicating that the variant creates a cryptic splicing site (AG). Our study has shed light on crucial phenotype–genotype correlations and provided valuable insights, particularly regarding the Latin American population.
期刊介绍:
Clinical Genetics links research to the clinic, translating advances in our understanding of the molecular basis of genetic disease for the practising clinical geneticist. The journal publishes high quality research papers, short reports, reviews and mini-reviews that connect medical genetics research with clinical practice.
Topics of particular interest are:
• Linking genetic variations to disease
• Genome rearrangements and disease
• Epigenetics and disease
• The translation of genotype to phenotype
• Genetics of complex disease
• Management/intervention of genetic diseases
• Novel therapies for genetic diseases
• Developmental biology, as it relates to clinical genetics
• Social science research on the psychological and behavioural aspects of living with or being at risk of genetic disease

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


