C–heteroatom coupling with electron-rich aryls enabled by nickel catalysis and light
IF 42.8
1区 化学
Q1 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Nickel photoredox catalysis has resulted in a rich development of transition-metal-catalysed transformations for carbon–heteroatom bond formation. By harnessing light energy, the transition metal can attain oxidation states that are difficult to achieve through thermal chemistry in a catalytic manifold. For example, nickel photoredox reactions have been reported for both the synthesis of anilines and aryl ethers from aryl(pseudo)halides. However, oxidative addition to simple nickel systems is often sluggish in the absence of special, electron-rich ligands, leading to catalyst decomposition. Electron-rich aryl electrophiles therefore currently fall outside the scope of many transformations in the field. Here we provide a conceptual solution to this problem and demonstrate nickel-catalysed C–heteroatom bond-forming reactions of arylthianthrenium salts, including amination, oxygenation, sulfuration and halogenation. Because the redox properties of arylthianthrenium salts are primarily dictated by the thianthrenium, oxidative addition of highly electron-rich aryl donors can be unlocked using simple NiCl2 under light irradiation to form the desired C‒heteroatom bonds. Photoredox-catalysed coupling of electron-rich aryl electrophiles based on simple nickel salts usually suffers from a slow oxidative addition. Now, it is shown that thianthrenation leads to more favourable redox properties of the substrates, alleviating this problem in carbon–heteroatom bond-forming reactions.
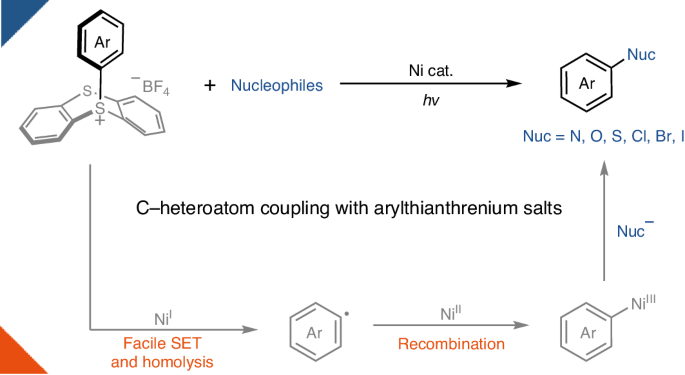
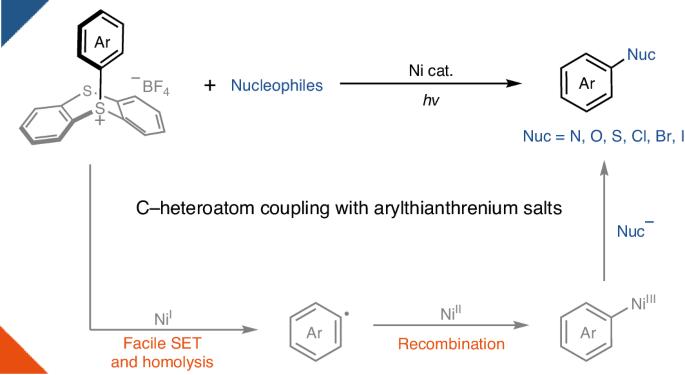
通过镍催化和光,实现 C-杂原子与富电子芳基的耦合
镍的光氧化催化技术为过渡金属催化碳-杂原子键形成的转化带来了丰富的发展。通过利用光能,过渡金属可以在催化歧管中达到热化学难以达到的氧化态。例如,有报道称镍光氧化反应可用于从芳基(假)卤化物合成苯胺和芳基醚。然而,在没有特殊的富电子配体的情况下,简单镍体系的氧化加成反应通常比较缓慢,从而导致催化剂分解。因此,富电子芳基亲电体目前不属于该领域许多转化的范围。在此,我们为这一问题提供了一个概念性解决方案,并演示了镍催化的芳基噻吩鎓盐的 C-杂原子键形成反应,包括胺化、氧化、硫化和卤化反应。由于芳基噻蒽盐的氧化还原特性主要由噻蒽决定,因此在光照射下使用简单的 NiCl2 就能解开高度富电子芳基供体的氧化添加,形成所需的 C-杂原子键。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Nature Catalysis
Chemical Engineering-Bioengineering
CiteScore
52.10
自引率
1.10%
发文量
140
期刊介绍:
Nature Catalysis serves as a platform for researchers across chemistry and related fields, focusing on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, encompassing both fundamental and applied studies. With a particular emphasis on advancing sustainable industries and processes, the journal provides comprehensive coverage of catalysis research, appealing to scientists, engineers, and researchers in academia and industry.
Maintaining the high standards of the Nature brand, Nature Catalysis boasts a dedicated team of professional editors, rigorous peer-review processes, and swift publication times, ensuring editorial independence and quality. The journal publishes work spanning heterogeneous catalysis, homogeneous catalysis, and biocatalysis, covering areas such as catalytic synthesis, mechanisms, characterization, computational studies, nanoparticle catalysis, electrocatalysis, photocatalysis, environmental catalysis, asymmetric catalysis, and various forms of organocatalysis.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


