KRAS/PI3K axis driven GTF3C6 expression and promotes LUAD via FAK pathway
IF 11.4
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
Abstract
Introduction
Effective targeting drugs for KRAS mutation-mediated Lung Adenocarcinoma (LUAD) are currently are limited.
Objectives
Investigating and intervening in the downstream key target genes of KRAS is crucial for clinically managing KRAS mutant-driven LUAD. GTF3C6, a newly identified member of the general transcription factor III (GTF3) family, plays a role in the transcription of RNA polymerase III (pol III)-dependent genes. However, its involvement in cancer remains unexplored.
Methods
This study examined the expression, roles, and potential molecular mechanisms of GTF3C6 in LUAD tissues, LSL-KrasG12D/+;LSL-p53−/− LUAD mouse models, and LUAD patients-derived organoid using Western blot, qRT-PCR, immunofluorescence, immunohistochemistry, and gene manipulation assays.
Results
We present the first evidence that GTF3C6 is highly expressed in LUAD tissues, LSL-KrasG12D/+;LSL-p53−/− LUAD mouse models, and LUAD organoids, correlating with poor clinical prognosis. Furthermore, GTF3C6 was found to promote anchorage-independent proliferation, migration, and invasion of LUAD cells. Mechanistically, KRAS mutation drives GTF3C6 expression through the PI3K pathway, and GTF3C6 knockdown reverses the malignant phenotype of KRAS mutation-driven LUAD cells. Additionally, the FAK pathway emerged as a crucial downstream signaling pathway through which GTF3C6 mediates the malignant phenotype of LUAD. Finally, GTF3C6 knockdown suppresses LUAD organoid formation and inhibits tumor growth in vivo.
Conclusion
Our findings demonstrate that GTF3C6, driven by KRAS mutation, promotes LUAD development by regulating FAK phosphorylation, suggesting its potential as a biomarker and therapeutic target in KRAS mutant-driven LUAD.

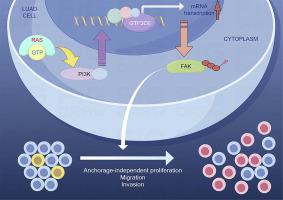
KRAS/PI3K 轴驱动 GTF3C6 的表达,并通过 FAK 途径促进 LUAD
目前,针对 KRAS 突变介导的肺腺癌(LUAD)的有效靶向药物非常有限。研究和干预 KRAS 下游的关键靶基因对于临床治疗 KRAS 突变驱动的肺腺癌至关重要。GTF3C6 是新发现的通用转录因子 III(GTF3)家族成员,在依赖 RNA 聚合酶 III(pol III)的基因转录中发挥作用。然而,它在癌症中的参与仍未得到探索。本研究采用 Western 印迹、qRT-PCR、免疫荧光、免疫组化和基因操作等方法,研究了 GTF3C6 在 LUAD 组织、LSL-Kras;LSL-p53 LUAD 小鼠模型和 LUAD 患者衍生的器官组织中的表达、作用和潜在的分子机制。我们首次证明了 GTF3C6 在 LUAD 组织、LSL-Kras;LSL-p53 LUAD 小鼠模型和 LUAD 器官组织中的高表达与不良临床预后相关。此外,研究还发现 GTF3C6 能促进 LUAD 细胞不依赖锚的增殖、迁移和侵袭。从机理上讲,KRAS突变通过PI3K通路驱动GTF3C6的表达,敲除GTF3C6可逆转KRAS突变驱动的LUAD细胞的恶性表型。此外,FAK通路也是GTF3C6介导LUAD恶性表型的重要下游信号通路。最后,敲除 GTF3C6 可抑制 LUAD 器官形成并抑制体内肿瘤生长。我们的研究结果表明,KRAS突变驱动的GTF3C6通过调节FAK磷酸化促进了LUAD的发展,这表明它有可能成为KRAS突变驱动的LUAD的生物标记物和治疗靶点。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of Advanced Research
Multidisciplinary-Multidisciplinary
CiteScore
21.60
自引率
0.90%
发文量
280
审稿时长
12 weeks
期刊介绍:
Journal of Advanced Research (J. Adv. Res.) is an applied/natural sciences, peer-reviewed journal that focuses on interdisciplinary research. The journal aims to contribute to applied research and knowledge worldwide through the publication of original and high-quality research articles in the fields of Medicine, Pharmaceutical Sciences, Dentistry, Physical Therapy, Veterinary Medicine, and Basic and Biological Sciences.
The following abstracting and indexing services cover the Journal of Advanced Research: PubMed/Medline, Essential Science Indicators, Web of Science, Scopus, PubMed Central, PubMed, Science Citation Index Expanded, Directory of Open Access Journals (DOAJ), and INSPEC.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


