Dilek Cavusoglu, Gulten Ozturk, Dilsad Turkdogan, Semra Hiz Kurul, Uluc Yis, Mustafa Komur, Faruk Incecik, Bulent Kara, Turkan Sahin, Olcay Unver, Cengiz Dilber, Gulen Gul Mert, Cagatay Gunay, Gamze Sarikaya Uzan, Ozlem Ersoy, Yavuz Oktay, Serdar Mermer, Gokcen Oz Tuncer, Olcay Gungor, Gul Demet Kaya Ozcora, Ugur Gumus, Ozlem Sezer, Gokhan Ozan Cetin, Fatma Demir, Arzu Yilmaz, Gurkan Gurbuz, Meral Topcu, Haluk Topaloglu, Ahmet Cevdet Ceylan, Serdar Ceylaner, Joseph G. Gleeson, Dilara Fusun Icagasioglu, F. Mujgan Sonmez
{"title":"Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study","authors":"Dilek Cavusoglu, Gulten Ozturk, Dilsad Turkdogan, Semra Hiz Kurul, Uluc Yis, Mustafa Komur, Faruk Incecik, Bulent Kara, Turkan Sahin, Olcay Unver, Cengiz Dilber, Gulen Gul Mert, Cagatay Gunay, Gamze Sarikaya Uzan, Ozlem Ersoy, Yavuz Oktay, Serdar Mermer, Gokcen Oz Tuncer, Olcay Gungor, Gul Demet Kaya Ozcora, Ugur Gumus, Ozlem Sezer, Gokhan Ozan Cetin, Fatma Demir, Arzu Yilmaz, Gurkan Gurbuz, Meral Topcu, Haluk Topaloglu, Ahmet Cevdet Ceylan, Serdar Ceylaner, Joseph G. Gleeson, Dilara Fusun Icagasioglu, F. Mujgan Sonmez","doi":"10.1007/s12311-024-01690-1","DOIUrl":null,"url":null,"abstract":"<p>Pontocerebellar hypoplasia (PCH) is a heterogeneous group of neurodegenerative disorders characterized by hypoplasia and degeneration of the cerebellum and pons. We aimed to identify the clinical, laboratory, and imaging findings of the patients with diagnosed PCH with confirmed genetic analysis. We collected available clinical data, laboratory, and imaging findings in our retrospective multicenter national study of 64 patients with PCH in Turkey. The genetic analysis included the whole-exome sequencing (WES), targeted next-generation sequencing (NGS), or single gene analysis. Sixty-four patients with PCH were 28 female (43.8%) and 36 (56.3%) male. The patients revealed homozygous mutation in 89.1%, consanguinity in 79.7%, pregnancy at term in 85.2%, microcephaly in 91.3%, psychomotor retardation in 98.4%, abnormal neurological findings in 100%, seizure in 63.8%, normal biochemistry and metabolic investigations in 92.2%, and dysmorphic findings in 51.2%. The missense mutation was found to be the most common variant type in all patients with PCH. It was detected as <i>CLP1</i> (<i>n</i> = 17) was the most common PCH related gene. The homozygous missense variant c.419G > A (p.Arg140His) was identified in all patients with <i>CLP1.</i> Moreover, all patients showed the same homozygous missense variant c.919G > T (p.A307S) in <i>TSEN54</i> group (<i>n</i> = 6). In Turkey, <i>CLP1</i> was identified as the most common causative gene with the identical variant c.419G > A; p.Arg140His. The current study supports that genotype data on PCH leads to phenotypic variability over a wide phenotypic spectrum.</p>","PeriodicalId":22415,"journal":{"name":"The Cerebellum","volume":"65 1","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Cerebellum","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s12311-024-01690-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
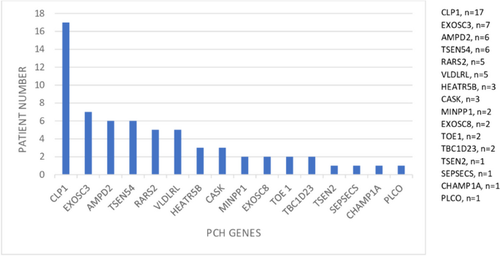
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of neurodegenerative disorders characterized by hypoplasia and degeneration of the cerebellum and pons. We aimed to identify the clinical, laboratory, and imaging findings of the patients with diagnosed PCH with confirmed genetic analysis. We collected available clinical data, laboratory, and imaging findings in our retrospective multicenter national study of 64 patients with PCH in Turkey. The genetic analysis included the whole-exome sequencing (WES), targeted next-generation sequencing (NGS), or single gene analysis. Sixty-four patients with PCH were 28 female (43.8%) and 36 (56.3%) male. The patients revealed homozygous mutation in 89.1%, consanguinity in 79.7%, pregnancy at term in 85.2%, microcephaly in 91.3%, psychomotor retardation in 98.4%, abnormal neurological findings in 100%, seizure in 63.8%, normal biochemistry and metabolic investigations in 92.2%, and dysmorphic findings in 51.2%. The missense mutation was found to be the most common variant type in all patients with PCH. It was detected as CLP1 (n = 17) was the most common PCH related gene. The homozygous missense variant c.419G > A (p.Arg140His) was identified in all patients with CLP1. Moreover, all patients showed the same homozygous missense variant c.919G > T (p.A307S) in TSEN54 group (n = 6). In Turkey, CLP1 was identified as the most common causative gene with the identical variant c.419G > A; p.Arg140His. The current study supports that genotype data on PCH leads to phenotypic variability over a wide phenotypic spectrum.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


