Amir M. Mofrad, Matthew S. Christian, Juliano Schorne-Pinto, Jorge P. S. Palma, Theodore M. Besmann
{"title":"Effect of XC functionals and dispersion corrections on the DFT-computed structural and vibrational properties of SrCl2–NaCl and ZrF4–LiF","authors":"Amir M. Mofrad, Matthew S. Christian, Juliano Schorne-Pinto, Jorge P. S. Palma, Theodore M. Besmann","doi":"10.1002/jrs.6670","DOIUrl":null,"url":null,"abstract":"<p>Density functional theory (DFT) calculations were performed to examine the impact of exchange–correlation (XC) functionals and van der Waals corrections (specifically the D3 method) on the structural and vibrational properties of the SrCl<sub>2</sub>–NaCl and ZrF<sub>4</sub>–LiF salt systems. Multiple XC functionals, including the local density approximation (LDA), the generalized gradient approximation using the Perdew–Burke–Ernzerhof (PBE) model, and its modified form suitable for solids (PBEsol), the dispersion-corrected PBE-D3 and PBEsol-D3, were considered. Of these functionals, LDA was found to exhibit the highest degree of error, while PBEsol and PBE-D3 displayed the least error. Underestimated lattice parameters compared with experimental values were observed to result in higher force constants, leading to an overprediction of vibrational frequencies. Conversely, an overestimation of lattice parameters was associated with lower vibrational frequencies. The methodology presented in this study yielded results that are in good agreement with experiment, irrespective of the method (finite differences vs. density functional perturbation theory) employed for calculating infrared and Raman spectra. It was further demonstrated that for alkali halides with weak Raman scattering, utilizing a supercell constructed from primitive cells better predicts Raman features than does the use of conventional cells.</p>","PeriodicalId":16926,"journal":{"name":"Journal of Raman Spectroscopy","volume":"55 7","pages":"819-832"},"PeriodicalIF":2.4000,"publicationDate":"2024-04-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jrs.6670","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Raman Spectroscopy","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jrs.6670","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"SPECTROSCOPY","Score":null,"Total":0}
引用次数: 0
Abstract
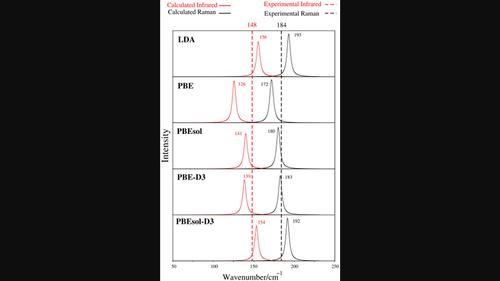
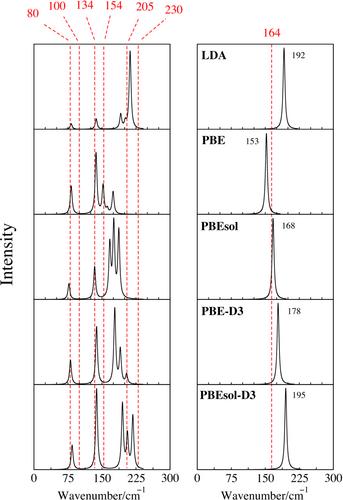
Density functional theory (DFT) calculations were performed to examine the impact of exchange–correlation (XC) functionals and van der Waals corrections (specifically the D3 method) on the structural and vibrational properties of the SrCl2–NaCl and ZrF4–LiF salt systems. Multiple XC functionals, including the local density approximation (LDA), the generalized gradient approximation using the Perdew–Burke–Ernzerhof (PBE) model, and its modified form suitable for solids (PBEsol), the dispersion-corrected PBE-D3 and PBEsol-D3, were considered. Of these functionals, LDA was found to exhibit the highest degree of error, while PBEsol and PBE-D3 displayed the least error. Underestimated lattice parameters compared with experimental values were observed to result in higher force constants, leading to an overprediction of vibrational frequencies. Conversely, an overestimation of lattice parameters was associated with lower vibrational frequencies. The methodology presented in this study yielded results that are in good agreement with experiment, irrespective of the method (finite differences vs. density functional perturbation theory) employed for calculating infrared and Raman spectra. It was further demonstrated that for alkali halides with weak Raman scattering, utilizing a supercell constructed from primitive cells better predicts Raman features than does the use of conventional cells.
期刊介绍:
The Journal of Raman Spectroscopy is an international journal dedicated to the publication of original research at the cutting edge of all areas of science and technology related to Raman spectroscopy. The journal seeks to be the central forum for documenting the evolution of the broadly-defined field of Raman spectroscopy that includes an increasing number of rapidly developing techniques and an ever-widening array of interdisciplinary applications.
Such topics include time-resolved, coherent and non-linear Raman spectroscopies, nanostructure-based surface-enhanced and tip-enhanced Raman spectroscopies of molecules, resonance Raman to investigate the structure-function relationships and dynamics of biological molecules, linear and nonlinear Raman imaging and microscopy, biomedical applications of Raman, theoretical formalism and advances in quantum computational methodology of all forms of Raman scattering, Raman spectroscopy in archaeology and art, advances in remote Raman sensing and industrial applications, and Raman optical activity of all classes of chiral molecules.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


